有害生物防除剂的制造法的制作方法
技术领域
本发明涉及具有2-酰基亚氨基吡啶结构的新的有害生物防除剂的制造方法。
现有技术
目前为止已发现多种有害生物防除剂,然而因为药剂灵敏性低下的问题、效果的持续性、使用时的安全性等,目前依然追求新的药剂。
尤其是在东亚、东南亚的水稻栽培中,如非专利文献1所示,由对包含以吡虫啉(Imidacloprid)为代表的新烟碱(Neonicotinoids)类、及以氟虫腈(Fipronil)为代表的苯基吡唑系药剂等的主要杀虫剂发展了药剂抵抗性的飞虱类造成的灾害已表面化,期待针对发展了抵抗性的飞虱类的特效药。另外,要求稳定且廉价地提供作为有害生物防除剂所要求的量的这些新的药剂。
作为具有2-酰基亚氨基吡啶结构的有害生物防除剂的制造方法,已知专利文献1~3、非专利文献2。专利文献1公开了具有与后述式(I)所表示的化合物同样的环结构的除草剂。专利文献2、以及专利文献3公开了具有与式(I)所表示的化合物类似的环结构的杀虫剂。非专利文献2公开了将与具有式(I)所表示的化合物类似的环结构的化合物作为合成中间体。
然而,专利文献1、专利文献2、专利文献3、非专利文献2所记载的制造方法,是将后述的式(Ba)所表示的化合物作为中间体的制造方法,没有记载将后述的式(B)所表示的化合物作为中间体的制造。另外,专利文献1、专利文献2、专利文献3、非专利文献2中公开了将式(Ba)所表示的化合物作为中间体的制造方法,但没有关于后述的式(Ia)所表示的化合物的制造的具体记载。另外,对于N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺,公开了结构式,作为该化合物的物性值记载了折射率为nD(25.5)=1.4818(专利文献2的第1表化合物号3),但不包含在已认定有害生物防除活性的化合物组的列表中(专利文献2的第2表、第3表)。
进而,专利文献3公开了N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的结构式,作为该化合物的物性值记载了熔点为60~62℃(专利文献3的表7的实施例号12),在实施例中并未举出具有害生物防除活性的化合物的例子。专利文献2及专利文献3中并未公开N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的具体制造方法。
另外,非专利文献3中公开了作为2-乙酰胺吡啶的互变异构体的N-(吡啶-2(1H)-亚基]-乙酰胺,但没有记载其具体制造方法、和/或卤代衍生物的制造方法。
现有技术文献
专利文献
专利文献1:欧洲专利申请公开第432600号说明书
专利文献2:日本特开平05-78323号公报
专利文献3:欧洲专利申请公开第268915号说明书
非专利文献
非专利文献1:Masaya Matsumura等,Pest Management Science,2008年、64卷、11号、1115~1121页
非专利文献2:Botho Kickhofen等,Chemische Berichte,1955年、88卷、1103~1108页
非专利文献3:Wladysl,aw Pietrzycki等,Bulletin des Societes Chimiques Belges,1993年、102卷、11-12号、709~717页
技术实现要素:
发明要解决的课题
提供后述式(I)所表示的具有2-酰基亚氨基吡啶结构的有害生物防除剂,特别是提供稳定且廉价地提供作为有害生物防除剂所要求的量的N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的制造方法。
用于解决课题的方法
即,根据第一发明,本发明者们通过以式(A)所表示的化合物作为出发物质,以式(B)所表示的化合物作为中间体,而得到作为目的有用的下述式(I)所表示的化合物,从而完成了本发明。
提供下述式(I)所表示的化合物的制造方法,
[式中,Ar表示可被取代的苯基、或可被取代的5~6元的杂环,R1表示可被取代的C1-6的烷基,Y表示氢原子、卤原子、羟基、可被卤原子取代的C1-6烷基、可被卤原子取代的C1-6烷氧基、氰基、甲酰基、硝基。]
该方法如下述反应式所示,包括:式(B)所表示的化合物的制造工序、和烷基化工序,
[式中,R1和Y表示与上述相同的含义,R2表示(1)三氟乙酰氧基、(2)可被卤原子取代的C1-6烷氧基、或苯基可被卤原子、甲基、氰基、硝基、甲氧基取代的苄氧基、(3)除了三氟乙酰氧基以外的可被卤原子取代的C1-6烷基羰氧基、苯基可被卤原子、甲基、氰基、硝基、甲氧基取代的苯基羰氧基、(4)羟基、(5)卤原子,R4表示卤原子、可被卤原子取代的C1-6烷基磺酰氧基(C1-6alkylsulfoxy group)、可被卤原子或甲基取代的苯基磺酰氧基(phenylsulfoxy group)。
所述式(B)所表示的化合物的制造工序是,将式(A)所表示的化合物的2位氨基,使用R1COR2所表示的酰基化剂进行酰基化,从而制造式(B)所表示的化合物的工序,
所述烷基化工序是,将式(B)所表示的化合物的1位氮原子进一步使用Ar-CH2-R4进行烷基化的工序。
根据第二发明,提供所述式(B)所表示的有用中间体(但是,R1为甲基或苯基且Y为氢原子的化合物除外)、及其盐。
根据第三发明,提供下述式(Ia)所表示的化合物的制造方法,
[式中,R3表示卤原子、氰基、硝基、三氟甲基,X表示碳原子或氮原子,R1a表示被卤素取代的C1-6烷基。]
该制造方法如下述反应式所示。
[其中,R1a、R4、R3及X表示与上述相同的含义,R2a表示(1)三氟乙酰氧基、(2)可被卤原子取代的C1-6烷氧基、或苯基可被卤原子、甲基、氰基、硝基、甲氧基取代的苄氧基、(3)除了三氟乙酰氧基以外的可被卤原子取代的C1-6烷基羰氧基、苯基可被卤原子、甲基、氰基、硝基、甲氧基取代的苯基羰氧基、(4)羟基、或(5)卤原子。]
根据第四发明,提供通过下述反应式所制造的式(I’)所表示的化合物。
[其中,式(I’)所表示的化合物是具有以下的(a)或(b)、或者(a)及(b)的物性的N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺。(a)在粉末X射线衍射中,至少在下述衍射角(2θ)具有衍射角峰。衍射角:8.6±0.2°、14.2±0.2°、17.5±0.2°、18.3±0.2°、19.7±0.2°、22.3±0.2°、30.9±0.2°、35.3±0.2°
(b)在差示扫描量热分析(DSC)中,熔点显示155~158℃。]
发明的效果
通过本发明,可以根据需要通过一锅(One-pot)有效且收率良好地制造作为有害生物防除剂有用的2-酰基亚氨基吡啶衍生物。
附图说明
图1是显示以第一制法调制的N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的晶体的粉末X射线晶体分析的结果的图。
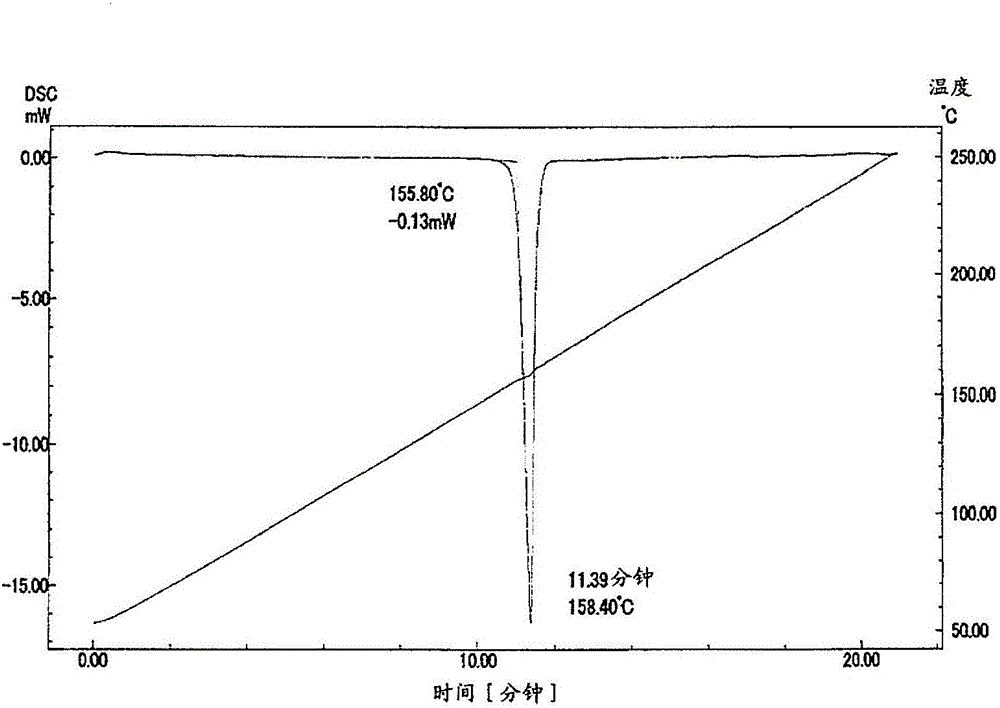
图2是显示以第一制法调制的N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的晶体的差示扫描量热分析的结果的图。
图3是显示以第二制法调制的N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的晶体的粉末X射线晶体分析的结果的图。
图4是显示以第二制法调制的N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的晶体的差示扫描量热分析的结果的图。
图5是显示以第三制法调制的N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的晶体的差示扫描量热分析的结果的图。
图6是显示以第四制法调制的N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的晶体的粉末X射线晶体分析的结果的图。
图7是显示以第四制法调制的N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的晶体的差示扫描量热分析的结果的图。
图8是显示以第五制法调制的N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的晶体的差示扫描量热分析的结果的图。
图9是显示合成例4中合成的N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的晶体的粉末X射线晶体分析的结果的图。
具体实施方式
在本说明书中,作为取代基或取代基的一部份的“烷基”这样的用语,只要没有特别定义,则分别是指直链状、支链状、环状或它们的组合的烷基。
在本说明书中,“卤原子”是指选自氟、氯、溴、碘中的原子。
在本说明书中,作为碱的“当量”,例如相对于1摩尔式(A)所表示的化合物使用1摩尔碳酸钾的情形时为2当量,使用1摩尔氢氧化钠或碳酸氢钠的情形时为1当量,使用1摩尔有机碱的情形时为1当量。
在本说明书中,“盐”表示盐酸盐、硫酸盐、硝酸盐等无机酸盐、三氟乙酸盐、二氟乙酸盐、二氯乙酸盐等有机酸盐等。
在本说明书中,R2表示羟基时,与酰基化剂R1COR2同时使用的“试剂”可以是其水合物。
在本说明书中,“缩合剂”表示N,N’-二环己基碳二亚胺、1-乙基-3-(3-二甲基氨基丙基)碳二亚胺盐酸盐、1,1’-羰基二咪唑、二吡啶基二硫、二咪唑基二硫、1,3,5-三氯苯甲酰氯、1,3,5-三氯苯甲酸酐、PyBop(注册商标,六氟磷酸(苯并三唑-1-基氧基)三吡咯烷基)、PyBrop(注册商标,六氟磷酸溴三(吡咯烷基))等用于合成酯和/或酰胺等羧酸衍生物的反应试剂。
在本说明书中,取代基所附带的“Ca-b”记号,是指该取代基所含有的碳原子数为a个~b个。另外,例如,在“Ca-b烷基羰基氧基”这种情形下的“Ca-b”,是指除了羰基氧基部分的碳原子之外,烷基部分的碳原子数为a个~b个。
Ar表示可被取代的苯基、或可被取代的5~6元的杂环,作为5~6元的杂环,可列举吡啶、嘧啶、噻唑、四氢呋喃、呋喃等,优选列举3-吡啶基、5-嘧啶基、3-噻唑基、5-噻唑基,更优选为3-吡啶基。作为可被导入到所述苯基或杂环中的取代基,可列举卤原子、可被卤原子取代的C1-4烷基、可被卤原子取代的C1-4烷氧基、羟基、氰基、硝基,优选为卤原子、可被卤原子取代的C1-4烷基,特别优选为氯原子。作为可被取代的苯基、及可被取代的5~6元的杂环,具体为苯基、3-氯苯基、4-氯苯基、3-氰基苯基、4-氰基苯基、3-硝基苯基、4-硝基苯基、3,5-二氯苯基、4-甲基苯基、4-甲氧基苯基、3,5-二溴苯基、2,4-二溴苯基、4-氟苯基、4-溴苯基、3-硝基-5-溴苯基、3,5-双三氟甲基苯基、6-氯-3-吡啶基、2-氯-5-噻唑基、6-氯-5-氟-3-吡啶基、6-溴-3-吡啶基、6-氟-3-吡啶基、5,6-二氯-3-吡啶基、6-三氟甲基-3-吡啶基,优选为6-氯-3-吡啶基、6-氟-3-吡啶基、6-氯-5-氟-3-吡啶基、6-溴-3-吡啶基,特别优选为6-氯-3-吡啶基。
R1表示可被取代的C1-6烷基,可被导入到所述C1-6烷基中的取代基可列举卤原子、C1-6卤代烷氧基、氰基、硝基、羟基,具体表示三氟甲基、二氟氯甲基、三氯甲基、五氟乙基、二氟甲基、二氯甲基、二溴甲基、氯甲基、二氟乙基、二氯乙基、2,2,2-三氟乙基、二氟环丙基、溴二氟甲基、三氟甲氧基甲基等,优选表示三氟甲基、二氟氯甲基、二氟甲基、三氯甲基、五氟乙基,更优选表示三氟甲基。
R1a表示可被卤素取代的C1-6烷基,可列举三氟甲基、三氯甲基、二氟氯甲基、二氟甲基、二氯甲基、二溴甲基、氯甲基、二氟乙基、二氯乙基、2,2,2-三氟乙基、五氟乙基、二氟环丙基等,优选为三氟甲基、三氯甲基、二氯甲基、二氟甲基、二氟氯甲基、氯甲基、五氟乙基,更优选为三氟甲基、二氟甲基、二氟氯甲基、氯甲基、五氟乙基,特别优选为三氟甲基。
Y表示氢原子、卤原子、羟基、可被卤原子取代的C1-6烷基、可被卤原子取代的C1-6烷氧基、氰基、甲酰基、硝基,优选表示氢原子、卤原子、羟基,更优选表示氢原子。
R2及R2a表示(1)三氟乙酰氧基、(2)可被卤原子取代的C1-6烷氧基、或苯基可被卤原子、甲基、氰基、硝基、甲氧基取代的苄氧基、(3)除了三氟乙酰氧基以外的可被卤原子取代的C1-6烷基羰氧基、苯基可被卤原子、甲基、氰基、硝基、甲氧基取代的苯基羰氧基、(4)羟基、或(5)卤原子。
R3表示在吡啶环或嘧啶环的碳原子上取代的取代基,明显其数在吡啶的情况下为0~4,在嘧啶的情况下为0~3。作为R3所表示的取代基,为卤原子、氰基、硝基、三氟甲基,可以各自相同也可以不同。
R4表示卤素、可被卤原子取代的C1-6烷基磺酰氧基、可被卤原子、甲基取代的苯基磺酰氧基。
作为式(I)及(Ia)所表示的化合物,优选为化合物号1:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺、化合物号2:N-[1-((6-氯-5-氟吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺、化合物号19:N-[1-((6-氟吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺、化合物号3:N-[1-((6-溴吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺、化合物号8:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2-二氟乙酰胺、化合物号4:2-氯-N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2-二氟乙酰胺、化合物号7:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,3,3,3-五氟丙酰胺、化合物号6:N-[1-((2-氯嘧啶-5-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺。
在式(I)及式(Ia)所表示的化合物中,作为特别优选的例子,是式(I’)所表示的化合物,即,具有以下(a)或(b)、或者(a)及(b)的物性的N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺(但是,除了专利文献2所记载的显示nD(25.5)=1.4818的化合物之外)。
(a)在粉末X射线衍射中,至少在下述衍射角(2θ)具有衍射角峰。衍射角:8.6±0.2°、14.2±0.2°、17.5±0.2°、18.3±0.2°、19.7±0.2°、22.3±0.2°、30.9±0.2°、35.3±0.2°
(b)在差示扫描量热分析(DSC)中,熔点显示155~158℃。
作为式(B)所表示的化合物的优选例,可列举2,2,2-三氟-N-(吡啶-2(1H)-亚基)乙酰胺、2-氯-2,2-二氟-N-(吡啶-2(1H)-亚基)乙酰胺、2,2,3,3,3-五氟-N-(吡啶-2(1H)-亚基)丙酰胺、2,2-二氟-N-(吡啶-2(1H)-亚基)乙酰胺,作为更优选的例子是下式(B1)所表示的2,2,2-三氟-N-(吡啶-2(1H)-亚基)乙酰胺。
制造方法
按照以下的流程图更详细地说明本发明。
[在上述的流程图中,Ar、Y、R1、R2、R4表示与上述相同的含义。]
另外,上述流程图所示的式(B)所表示的化合物可以不进行后处理或分离地在之后的工序中使用。
1-1:从式(A)所表示的化合物制造式(B)所表示的化合物
式(A)所表示的化合物可以是市售的化合物或通过例如Journal of labeled compounds&radiopharmaceuticals(1987),24(2)119-123所记载的方法获得。
作为从式(A)所表示的化合物制造式(B)所表示的化合物的方法,可以通过在无溶剂条件下或在不影响反应的溶剂中,使式(A)所表示的化合物,在碱存在下或不存在下,与酰基化剂R1COR2(R1、R2表示与上述所定义的相同的含义)反应而得到。
这里所说的试剂的当量数是相对于全部式(A)所表示的化合物的当量数。
作为可用的溶剂,可列举甲苯、二甲苯、乙苯等芳香族烃系溶剂、乙酸乙酯、乙酸丁酯等酯系溶剂、乙醚、异丙醚、四氢呋喃、二烷等醚系溶剂、N,N-二甲基甲酰胺、二甲基亚砜、N,N-二甲基乙酰胺、N-甲基-2-吡咯烷酮、乙腈等非质子性极性有机溶剂、二氯甲烷、氯仿等卤素系溶剂、环己烷等烃系溶剂、丙酮、甲乙酮等酮系溶剂、水、或它们的混合溶剂。
作为可用的碱,可列举碳酸钠、碳酸钾、碳酸氢钠、碳酸氢钾、氢氧化钠、氢氧化镁、氢氧化钙、氢氧化锂、氢氧化钡等无机碱、1,8-二氮杂二环[5.4.0]十一碳-7-烯、1,5-二氮杂二环[4.3.0]壬-5-烯、三乙胺、二异丙基乙基胺、吡啶、甲基吡啶、二甲基氨基吡啶等有机碱、乙醇钠、甲醇钠、叔丁醇钾等醇化物。碱也可以不使用,但在碱存在下进行时的使用量,可用0.01~20.0当量。
作为酰基化剂R1COR2,可列举三氟乙酸酐、三氟乙酸、三氟乙酸乙酯、三氟乙酰氯、或混合酸酐。另外,这样的酰基化剂可以单独使用或2种以上混合使用。其中,优选可使用三氟乙酸酐、三氟乙酸、三氟乙酸乙酯、或三氟乙酰氯。另外,R2表示羟基时,可以通过将N,N’-二环己基碳二亚胺、1-乙基-3-(3-二甲基氨基丙基)碳二亚胺盐酸盐、1,1’-羰基二咪唑、二吡啶基二硫、二咪唑基二硫、1,3,5-三氯苯甲酰氯、1,3,5-三氯苯甲酸酐、PyBop(注册商标,六氟磷酸(苯并三唑-1-基氧基)三吡咯烷基)、PyBrop(注册商标,六氟磷酸溴三(吡咯烷基))等缩合剂、五氧化二磷、硫酸、多聚磷酸、亚硫酰氯、三氯氧磷、乙二酰氯、三氟化硼、对甲苯磺酸、或铁、钴、铜、镍、锌、铝、锂或镁的卤化物、硫酸盐、硝酸盐或氧化物等试剂同时使用来进行。另外,这些试剂可单独使用或2种以上组合使用。作为铁、钴、铜、镍、锌、铝、锂或镁的卤化物、硫酸盐、硝酸盐或氧化物的优选例,可列举氯化锌、氯化铜、氯化镁、氯化钴、氯化镍、氯化亚铁、氯化铝、硫酸铁、硫酸铝。另外,这些金属化合物可以是其无水合物,也可以是水合物。酰基化剂的使用量优选为0.5~10.0当量,更优选为1.0~5.0当量。
反应温度优选为-80℃~200℃的范围。反应时间优选为0.1小时~7天的范围。
作为优选的方式,
(1)在R2表示三氟乙酰氧基时,具体地在作为酰基化剂使用三氟乙酸酐时,作为优选的溶剂,可列举乙酸乙酯、乙酸丁酯等酯系溶剂、二氯甲烷、氯仿等卤素系溶剂、甲苯、二甲苯、乙苯等芳香族烃系溶剂,更优选为甲苯。反应优选在碱不存在下进行,但在使用碱的情况下,作为优选的碱,可列举碳酸钠、碳酸钾、碳酸氢钾、三乙胺、吡啶等,更优选为碳酸钾。酰基化剂的使用量优选为1.0~5.0当量,更优选为1.0~1.5当量。使用碱时的碱使用量优选为1.0~4.5当量,更优选为1.0~3.0当量。反应温度优选为-20℃~50℃的范围,更优选为-10℃~30℃。反应时间优选为0.1小时~7天的范围,更优选为0.5小时~4小时的范围。特别优选的条件是,作为酰基化剂使用三氟乙酸酐,作为溶剂使用甲苯,酰基化剂的使用量为1.0~1.5当量,反应温度为-10℃~30℃,反应时间为0.5~4小时的条件。碱不存在下,或在使用碱的情况下是使用碳酸钾、其当量数使用1.0~3.0当量的条件。
(2)在R2表示可被卤原子取代的C1-6烷氧基、或苯基可被卤原子、甲基、氰基、硝基、甲氧基取代的苄氧基时,具体地在使用三氟乙酸乙酯、三氟乙酸甲酯、三氟乙酸丙酯、特别优选为三氟乙酸乙酯等时,作为优选的溶剂,为N,N-二甲基甲酰胺、二甲基亚砜、N,N-二甲基乙酰胺、N-甲基-2-吡咯烷酮、乙腈等非质子性极性有机溶剂、乙醚、异丙醚、四氢呋喃、二烷等醚系溶剂、及这些溶剂与甲苯、二甲苯、乙苯等芳香族烃系溶剂的混合溶剂,优选为N,N-二甲基甲酰胺、或N,N-二甲基甲酰胺与甲苯的混合溶剂。反应优选在碱不存在下进行,但在使用碱的情况下,作为优选的碱,可列举碳酸钾、三乙胺、二甲基氨基吡啶等,更优选为碳酸钾、二甲基氨基吡啶。酰基化剂的使用量优选为1.0~5.0当量,更优选为1.5~5.0当量。使用碱时的碱使用量优选为0.01~3.0当量,更优选为0.01~2.0当量。反应温度优选为20℃~100℃的范围,更优选为40℃~80℃。反应时间优选为0.1小时~7天的范围,更优选为1小时~2天的范围。
特别优选的条件是,作为酰基化剂使用三氟乙酸乙酯,作为溶剂使用N,N-二甲基甲酰胺、或N,N-二甲基甲酰胺与甲苯的混合溶剂,酰基化剂的使用量为1.5~5.0当量,反应温度为40℃~80℃,反应时间为2小时~2天的条件。碱不存在下,或在使用碱的情况下是使用碳酸钾、其当量数使用1.0~3.0当量的条件。
(3)在R2表示可被卤原子取代的C1-6烷基羰氧基(但是,除了三氟乙酰氧基以外)、苯基可被卤原子、甲基、氰基、硝基、甲氧基取代的苯基羰氧基时,具体可列举三甲基乙酰基。反应温度优选为-20℃~50℃的范围,更优选为-10℃~30℃。反应时间优选为0.1小时~7天的范围,更优选为0.5小时~4小时的范围。
(4)在R2表示羟基时,作为具体的酰基化剂可列举三氟乙酸、二氟氯乙酸、三氯乙酸、二氟乙酸、二氯乙酸、二溴乙酸、氯乙酸、二氟丙酸、二氯丙酸、2,2,2-三氟丙酸、五氟丙酸、二氟环丙烷甲酸等,优选为三氟乙酸、三氯乙酸、二氯乙酸、二氟乙酸、二氟氯乙酸、氯乙酸、五氟丙酸,更优选为三氟乙酸、二氟乙酸、二氟氯乙酸、五氟丙酸,特别优选为三氟乙酸。使用三氟乙酸时,作为优选的溶剂,可列举甲苯、二甲苯、乙苯等芳香族烃系溶剂及N,N-二甲基甲酰胺、二甲基亚砜、N,N-二甲基乙酰胺、N-甲基-2-吡咯烷酮、乙腈等非质子性极性有机溶剂,更优选为甲苯、二甲苯、N,N-二甲基甲酰胺、N-甲基-2-吡咯烷酮、N,N-二甲基乙酰胺、甲苯与N,N-二甲基甲酰胺的混合溶剂、二甲苯与N,N-二甲基甲酰胺的混合溶剂、二甲苯与N-甲基-2-吡咯烷酮的混合溶剂或二甲苯与N,N-二甲基乙酰胺的混合溶剂。作为同时使用的试剂,可列举N,N’-二环己基碳二亚胺、1-乙基-3-(3-二甲基氨基丙基)碳二亚胺盐酸盐、五氧化二磷、硫酸、多聚磷酸、亚硫酰氯、三氯氧磷、乙二酰氯等,这些溶剂优选使用0.2~5.0当量。另外,在作为同时使用的试剂使用氯化锌、氯化铜、氯化镁、氯化钴、氯化镍、氯化亚铁、氯化铝、硫酸铁、硫酸铝、三氟化硼、对甲苯磺酸等时,优选使用0.0001~1.0当量。反应使用五氧化二磷、硫酸、多聚磷酸、亚硫酰氯、三氯氧磷、乙二酰氯、氯化锌、氯化铜、氯化镁、氯化钴、氯化镍、氯化亚铁、氯化铝、硫酸铁、硫酸铝、三氟化硼、对甲苯磺酸时,优选在碱不存在下进行,使用N,N’-二环己基碳二亚胺、1-乙基-3-(3-二甲基氨基丙基)碳二亚胺盐酸盐时,优选在碱存在下进行。使用碱的情况下,作为优选的碱,可列举碳酸钠、碳酸钾、碳酸氢钾、三乙胺、吡啶、二甲基氨基吡啶等,更优选为三乙胺。酰基化剂的使用量优选为1.0~5.0当量,更优选为1.0~3.0当量。使用亚硫酰氯、三氯氧磷、乙二酰氯时,这些试剂使用0.2~5.0当量,反应温度优选为-30℃~80℃的范围,更优选为-10℃~40℃。使用五氧化二磷、硫酸、多聚磷酸时,这些试剂使用0.2~5.0当量,反应温度优选为-30℃~200℃的范围,更优选为-10℃~160℃。使用N,N’-二环己基碳二亚胺、1-乙基-3-(3-二甲基氨基丙基)碳二亚胺盐酸盐时,优选为这些试剂使用0.2~5.0当量,反应温度优选为-30℃~80℃的范围,更优选为-10℃~40℃,以三乙胺作为碱,使用0.2~5.0当量的条件。使用氯化锌、氯化铜、氯化镁、氯化钴、氯化镍、氯化亚铁、氯化铝、硫酸铁、硫酸铝、三氟化硼、对甲苯磺酸时,这些试剂使用0.0001~1.0当量,反应温度优选为20℃~200℃的范围,更优选为80℃~160℃。反应时间优选为0.1小时~7天的范围,更优选为0.5小时~2天的范围。
特别优选的条件是,作为酰基化剂使用三氟乙酸,作为溶剂使用甲苯、N,N-二甲基甲酰胺、二甲苯、N-甲基-2-吡咯烷酮、N,N-二甲基乙酰胺、N,N-二甲基甲酰胺与甲苯的混合溶剂、N,N-二甲基甲酰胺与二甲苯的混合溶剂、二甲苯与N-甲基-2-吡咯烷酮的混合溶剂或二甲苯与N,N-二甲基乙酰胺的混合溶剂,酰基化剂的使用量为1.0~3.0当量。使用亚硫酰氯、三氯氧磷、乙二酰氯时的特别优选的条件是,这些试剂使用0.3~3.0当量,在碱不存在下,反应温度为-10℃~40℃,反应时间为0.5小时~1天的条件。使用五氧化二磷、硫酸、多聚磷酸时的特别优选的条件是,这些试剂使用0.2~2.0当量,反应温度为-10℃~160℃,反应时间为0.5小时~1天的条件。使用N,N’-二环己基碳二亚胺、1-乙基-3-(3-二甲基氨基丙基)碳二亚胺盐酸盐时的特别优选的条件是,这些试剂使用0.5~3当量,反应温度为-10℃~40℃,以三乙胺作为碱,使用0.5~3.0当量,反应时间为0.5~1天的条件。使用氯化锌、氯化铜、氯化镁、氯化钴、氯化镍、氯化亚铁、氯化铝、硫酸铁、硫酸铝、三氟化硼、对甲苯磺酸时的特别优选的条件是,这些试剂使用0.0001~0.5当量,在碱不存在下,反应温度为80℃~160℃,反应时间为2小时~2天的条件。
(5)在R2表示卤原子时,具体在使用三氟乙酰氯、三氟乙酰溴,优选为三氟乙酰氯时,作为优选的溶剂,可列举氯仿、二氯甲烷等的卤素系溶剂、甲苯、二甲苯、乙苯等芳香族烃系溶剂、及N,N-二甲基甲酰胺、二甲基亚砜、N,N-二甲基乙酰胺、N-甲基-2-吡咯烷酮、乙腈等非质子性极性有机溶剂,更优选为甲苯、N,N-二甲基甲酰胺、N-甲基-2-吡咯烷酮、或它们的混合溶剂。反应优选在碱不存在下进行,但在使用碱的情况下,作为优选的碱,可列举碳酸钠、碳酸钾、碳酸氢钾、三乙胺、吡啶等,更优选为碳酸钾。酰基化剂的使用量优选为1.0~5.0当量,更优选为1.0~3.0当量。使用碱时的碱使用量优选为1.0~5.0当量,更优选为1.0~3.0当量。反应温度优选为-80℃~40℃的范围,更优选为-30℃~30℃。反应时间优选为0.1小时~7天的范围,更优选为0.5小时~8小时的范围。另外,R2表示氯原子时,还可以使用将三氟乙酸与亚硫酰氯、三氯氧磷、乙二酰氯等,在与式(A)所表示的化合物反应的体系外同时使用从而预先生成的R1COCl。
特别优选的条件是,作为酰基化剂使用三氟乙酰氯,作为溶剂使用甲苯、N,N-二甲基甲酰胺、N-甲基-2-吡咯烷酮或它们的混合溶剂,酰基化剂的使用量为1.0~3.0当量,反应温度为-30℃~30℃,反应时间为0.5小时~8小时的条件。特别优选碱不存在下,或在使用碱的情况下使用碳酸钾、其当量数使用1.0~3.0当量的条件。
在从式(A)所表示的化合物合成式(B)所表示的化合物后,可使用碱进行中和。作为可用的碱,可列举碳酸钠、碳酸钾、碳酸氢钠、碳酸氢钾、氢氧化钠、氢氧化镁、氢氧化钙、氢氧化锂、氢氧化钡等无机碱、1,8-二氮杂二环[5.4.0]十一碳-7-烯、1,5-二氮杂二环[4.3.0]壬-5-烯、三乙胺、二异丙基乙基胺、吡啶、甲基吡啶、二甲基氨基吡啶等有机碱、乙醇钠、甲醇钠、叔丁醇钾等醇化物,优选为碳酸钾、乙醇钠、三乙胺。
1-2:从式(B)或式(B’)所表示的化合物制造式(I)或式(I’)所表示的化合物
作为从式(B)或式(B’)所表示的化合物制造式(I)或式(I’)所表示的化合物的方法,可以通过在无溶剂条件下或在不影响反应的溶剂中,使式(B)或式(B’)所表示的化合物在碱存在下与Ar-CH2-R4(Ar、R4表示与上述所定义的相同的含义)反应而得到。
作为可用的溶剂,可列举乙醚、异丙醚、四氢呋喃、二烷等醚系溶剂、N,N-二甲基甲酰胺、二甲基亚砜、N,N-二甲基乙酰胺、乙腈、N-甲基-2-吡咯烷酮、N-甲基-2-哌嗪酮、N,N-二甲基-2-咪唑烷酮、乙腈等非质子性极性有机溶剂、二氯甲烷、氯仿等卤素系溶剂、甲苯、二甲苯、乙苯等芳香族烃系溶剂、及它们的混合溶剂,优选列举非质子性极性有机溶剂。更优选为N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、N-甲基-2-吡咯烷酮、N,N-二甲基-2-咪唑烷酮、乙腈,或N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、N-甲基-2-吡咯烷酮、N,N-二甲基-2-咪唑烷酮、乙腈与芳香族烃系溶剂的混合溶剂,特别优选为N,N-二甲基甲酰胺、或N,N-二甲基甲酰胺与甲苯的混合溶剂。
作为在碱存在下进行时的可用的碱,可列举碳酸钠、碳酸钾、碳酸氢钠、碳酸氢钾、氢氧化钠、氢氧化镁、氢氧化钙、氢氧化锂、氢氧化钡等无机碱、1,8-二氮杂二环[5.4.0]十一碳-7-烯、1,5-二氮杂二环[4.3.0]壬-5-烯、三乙胺、二异丙基乙基胺、吡啶、二甲基吡啶、可力丁、N,N-二甲基苯胺、N,N-二乙基苯胺等有机碱,优选列举碳酸钾、碳酸氢钾、吡啶、三乙胺等,更优选列举碳酸钾、三乙胺。
Ar-CH2-R4(Ar、R4表示与上述所定义的相同的含义)的使用量优选相对于式(B)或式(B’)所表示的化合物为0.7~2.0当量,更优选为0.8~1.5当量。使用碱时的碱使用量优选相对于式(B)或式(B’)所表示的化合物为1.0~10.0当量,更优选为1.0~5.0当量。
反应温度优选为20℃~100℃的范围,更优选为40℃~80℃。反应时间优选为0.1小时~3天的范围,更优选为1小时~2天的范围。
特别优选的条件是,R4为氯原子,作为溶剂使用N,N-二甲基甲酰胺、N-甲基-2-吡咯烷酮、N,N-二甲基乙酰胺、N,N-二甲基甲酰胺与甲苯的混合溶剂、N,N-二甲基甲酰胺与二甲苯的混合溶剂、二甲苯与N-甲基-2-吡咯烷酮的混合溶剂或二甲苯与N,N-二甲基乙酰胺的混合溶剂,Ar-CH2-R4的使用量相对于式(B)或式(B’)所表示的化合物为0.8~1.5当量,反应温度为40℃~80℃,反应时间为1小时~2天的条件,碱使用1.0~5.0当量的碳酸钾或三乙胺的条件。
从式(A)或式(A’)所表示的化合物经由式(B)或(B’)所表示的化合物得到式(I)或式(I’)所表示的化合物的一锅制造
从式(A)或式(A’)所表示的化合物合成式(I)或式(I’)所表示的化合物时,可以在不分离式B或式(B’)所表示的化合物地进行之后的工序,从而得到式(I)或式(I’)所表示的化合物。
具体而言,将式(B)或式(B’)所表示的反应物直接、或减压下除去过量的试剂后,添加Ar-CH2-R4(Ar、R4表示与上述相同的含义)和碱,在上述条件下反应,得到式(I)或式(I’)所表示的化合物。
作为从式(A)或式(A’)所表示的化合物经由式(B)或(B’)所表示的化合物得到式(I)或式(I’)所表示的化合物的方法的优选例,是在碱不存在下,使用芳香族烃系溶剂或非质子性极性溶剂、或其混合溶剂,使式(A)或式(A’)所表示的化合物与酰基化剂R1COR2反应而得到式(B)或式(B’)所表示的化合物后,加入芳香族烃系溶剂、或非质子性极性有机溶剂、或它们的混合溶剂、Ar-CH2-R4和碱,直接或在减压下馏去芳香族烃系溶剂的同时进行反应,得到式(I)或式(I’)所表示的化合物的方法。
在一锅制造中从式(A)或式(A’)所表示的化合物制造式(B)或(B’)所表示的化合物
这里所说的试剂的当量数,是相对于全部式(A)或式(A’)所表示的化合物的当量数。为了从式(A)或式(A’)所表示的化合物得到式(B)或式(B’)所表示的化合物,特别优选使用R2为CF3COO基、OEt基、羟基或氯原子的R1COR2、或CF3COR2。
R2为CF3COO基(例如,三氟乙酸酐)时,是作为溶剂使用甲苯,酰基化剂的使用量为1.0~1.5当量,反应温度为-10℃~30℃,反应时间为0.5~4小时的条件,特别优选碱不存在下,或在使用碱的情况下使用碳酸钾、其当量数使用1.0~3.0当量的条件。R2为OEt基(三氟乙酸乙酯)时,是作为溶剂使用N,N-二甲基甲酰胺、或N,N-二甲基甲酰胺与甲苯的混合溶剂,酰基化剂的使用量为1.5~5.0当量,反应温度为40~80℃,反应时间为2小时~2天的条件,特别优选在碱不存在下,或在使用碱的情况下使用碳酸钾或二甲基氨基吡啶、其当量数使用0.01~2.0当量的条件。
R2为羟基(例如,三氟乙酸)时的特别优选的条件是,作为溶剂使用甲苯、N,N-二甲基甲酰胺、二甲苯、N-甲基-2-吡咯烷酮、N,N-二甲基乙酰胺、N,N-二甲基甲酰胺与甲苯的混合溶剂、N,N-二甲基甲酰胺与二甲苯的混合溶剂、二甲苯与N-甲基-2-吡咯烷酮的混合溶剂或二甲苯与N,N-二甲基乙酰胺的混合溶剂,酰基化剂的使用量为1.0~3.0当量。使用亚硫酰氯、三氯氧磷、乙二酰氯时的特别优选的条件是,这些试剂使用0.3~3.0当量,在碱不存在下,反应温度为-10℃~40℃,反应时间为0.5小时~1天的条件。使用五氧化二磷、硫酸、多聚磷酸时的特别优选的条件是,这些试剂使用0.5~2.0当量,反应温度为-10℃~160℃,反应时间为0.5小时~1天的条件。使用N,N’-二环己基碳二亚胺、1-乙基-3-(3-二甲基氨基丙基)碳二亚胺盐酸盐时的特别优选的条件是,这些试剂使用0.5~3.0当量,反应温度为-10℃~40℃,以三乙胺作为碱使用0.5~3.0当量,反应时间为0.5~1天的条件。使用氯化锌、氯化铜、氯化镁、氯化钴、氯化镍、氯化亚铁、氯化铝、硫酸铁、硫酸铝、三氟化硼、对甲苯磺酸时的特别优选的条件是,这些试剂使用0.0001~0.5当量,在碱不存在下,反应温度为80℃~160℃,反应时间为2小时~2天的条件。
R2为氯原子(例如,三氟乙酰氯)时,是作为溶剂使用甲苯、N,N-二甲基甲酰胺、N-甲基-2-吡咯烷酮或它们的混合溶剂,酰基化剂的使用量为1.0~3.0当量,反应温度为-30℃~30℃、反应时间为0.5小时~8小时的条件。特别优选在碱不存在下,或使用碱的情况下使用碳酸钾、其当量数使用1.0~3.0当量的条件。
在一锅制造中从式(B)或式(B’)所表示的化合物制造式(I)或(I’)所表示的化合物
用于从式(B)或式(B’)所表示的化合物得到式(I)或式(I’)所表示的化合物的特别优选的条件是,R4为氯原子,作为溶剂使用N,N-二甲基甲酰胺、N-甲基-2-吡咯烷酮、N,N-二甲基乙酰胺、N,N-二甲基甲酰胺与甲苯的混合溶剂、N,N-二甲基甲酰胺与二甲苯的混合溶剂、二甲苯与N-甲基-2-吡咯烷酮的混合溶剂或二甲苯与N,N-二甲基乙酰胺的混合溶剂,Ar-CH2-R4的使用量相对于式(B)或式(B’)所表示的化合物为0.8~1.5当量,反应温度为40℃~80℃,反应时间为1小时~2天的条件,是碱使用1.0~5.0当量的碳酸钾或三乙胺的条件。
从式(Ba)所表示的化合物制造式(Ia)所表示的化合物的工序
作为从式(Ba)所表示的化合物得到式(Ia)所表示的化合物的方法,可以在无溶剂条件下或在不影响反应的溶剂中,使式(Ba)所表示的化合物在碱存在下或不存在下,与酰基化剂R1aCOR2a(R1a、R2a表示与上述所定义的相同的含义)反应,从而得到。这里所说的试剂的当量数,是相对于全部式(Ba)所表示的化合物的当量数。
作为可用的溶剂,可列举甲苯、二甲苯、乙苯等芳香族烃系溶剂、乙酸乙酯、乙酸丁酯等酯系溶剂、乙醚、异丙醚、四氢呋喃、二烷等醚系溶剂、N,N-二甲基甲酰胺、二甲基亚砜、N,N-二甲基乙酰胺、N-甲基-2-吡咯烷酮、乙腈等非质子性极性有机溶剂、二氯甲烷、氯仿等卤素系溶剂、环己烷等烃系溶剂、丙酮、甲乙酮等酮系溶剂、水、或它们的混合溶剂。
作为可用的碱,可列举碳酸钠、碳酸钾、碳酸氢钠、碳酸氢钾、氢氧化钠、氢氧化镁、氢氧化钙、氢氧化锂、氢氧化钡等无机碱、1,8-二氮杂二环[5.4.0]十一碳-7-烯、1,5-二氮杂二环[4.3.0]壬-5-烯、三乙胺、二异丙基乙基胺、吡啶、甲基吡啶、二甲基氨基吡啶等有机碱、乙醇钠、甲醇钠、叔丁醇钾等醇化物。可以不使用碱,但在碱存在下进行时的使用量可使用0.01~20.0当量。
作为酰基化剂R1COR2,可列举三氟乙酸酐、三氟乙酸、三氟乙酸乙酯、三氟乙酰氯、或混合酸酐。另外,这样的酰基化剂可以单独使用或2种以上混合使用。其中,优选可使用三氟乙酸酐、三氟乙酸、三氟乙酸乙酯、或三氟乙酰氯。另外,R2表示羟基时,可以通过将N,N’-二环己基碳二亚胺、1-乙基-3-(3-二甲基氨基丙基)碳二亚胺盐酸盐、1,1’-羰基二咪唑、二吡啶基二硫、二咪唑基二硫、1,3,5-三氯苯甲酰氯、1,3,5-三氯苯甲酸酐、PyBop(注册商标)、PyBrop(注册商标)、五氧化二磷、硫酸、多聚磷酸、亚硫酰氯、三氯氧磷、乙二酰氯、氯化锌、氯化铜、氯化镁、氯化钴、氯化镍、氯化亚铁、氯化铝、硫酸铁、硫酸铝、三氟化硼、对甲苯磺酸等试剂同时使用而进行。酰基化剂的使用量优选为0.5~10.0当量。
反应温度优选为-80℃~200℃的范围。反应时间优选为0.1小时~7天的范围。
作为优选的方式,
(1)在R2表示三氟乙酰氧基时,具体地在作为酰基化剂使用三氟乙酸酐时,作为优选的溶剂,可列举乙酸乙酯、乙酸丁酯等酯系溶剂、二氯甲烷、氯仿等卤素系溶剂、甲苯、二甲苯、乙苯等芳香族烃系溶剂,更优选为甲苯。反应优选在碱不存在下进行,但在使用碱的情况下,作为优选的碱,可列举碳酸钠、碳酸钾、碳酸氢钾、三乙胺、吡啶等,更优选为碳酸钾。酰基化剂的使用量优选为1.0~5.0当量,更优选为1.0~1.5当量。使用碱时的碱使用量优选为1.0~4.5当量,更优选为1.0~3.0当量。反应温度优选为-20℃~50℃的范围,更优选为-10℃~30℃。反应时间优选为0.1小时~7天的范围,更优选为0.5小时~4小时的范围。
特别优选的条件是,作为酰基化剂使用三氟乙酸酐,作为溶剂使用甲苯,酰基化剂的使用量为1.0~1.5当量,反应温度为-10℃~30℃,反应时间为0.5~4小时的条件。在碱不存在下,或在使用碱的情况下使用碳酸钾、其当量数使用1.0~3.0当量的条件。
(2)在R2表示可被卤原子取代的C1-6烷氧基、或苯基可被卤原子、甲基、氰基、硝基、甲氧基取代的苄氧基时,具体在使用三氟乙酸乙酯、三氟乙酸甲酯、三氟乙酸丙酯、特别优选为三氟乙酸乙酯等时,作为优选的溶剂,是N,N-二甲基甲酰胺、二甲基亚砜、N,N-二甲基乙酰胺、N-甲基-2-吡咯烷酮、乙腈等非质子性极性有机溶剂、乙醚、异丙醚、四氢呋喃、二烷等醚系溶剂、及这些的溶剂与甲苯、二甲苯、乙苯等芳香族烃系溶剂的混合溶剂,更优选为N,N-二甲基甲酰胺、或N,N-二甲基甲酰胺与甲苯的混合溶剂。反应优选在碱不存在下进行,但在使用碱的情况下,作为优选的碱,可列举碳酸钾、三乙胺、二甲基氨基吡啶等,更优选为碳酸钾、二甲基氨基吡啶。酰基化剂的使用量优选为1.0~5.0当量,更优选为1.5~5.0当量。使用碱时的碱使用量优选为0.01~3.0当量,更优选为0.01~2.0当量。反应温度优选为20℃~100℃的范围,更优选为40℃~80℃。反应时间优选为0.1小时~7天的范围,更优选为1小时~2天的范围。
特别优选的条件是,作为酰基化剂使用三氟乙酸乙酯,作为溶剂使用N,N-二甲基甲酰胺、或N,N-二甲基甲酰胺与甲苯的混合溶剂,酰基化剂的使用量为1.0~5.0当量、反应温度为40℃~80℃,反应时间为2小时~2天的条件。是在碱不存在下,或在使用碱的情况下使用碳酸钾或二甲基氨基吡啶、其当量数使用0.01~2.0当量的条件。
(3)在R2表示可被卤原子取代的C1-6烷基羰氧基(但是,除了三氟乙酰氧基以外)、苯基可被卤原子、甲基、氰基、硝基、甲氧基取代的苯基羰氧基时,具体可列举三甲基乙酰基。反应温度优选为-20℃~50℃的范围,更优选为-10℃~30℃。反应时间优选为0.1小时~7天的范围,更优选为0.5小时~4小时的范围。
(4)在R2表示羟基时,作为具体的酰基化剂可列举三氟乙酸、二氟氯乙酸、三氯乙酸、二氟乙酸、二氯乙酸、二溴乙酸、氯乙酸、二氟丙酸、二氯丙酸、2,2,2-三氟丙酸、五氟丙酸、二氟环丙烷甲酸等,优选为三氟乙酸、三氯乙酸、二氯乙酸、二氟乙酸、二氟氯乙酸、氯乙酸、五氟丙酸,更优选为三氟乙酸、二氟乙酸、二氟氯乙酸、五氟丙酸,特别优选为三氟乙酸。使用三氟乙酸时,作为优选的溶剂,可列举甲苯、二甲苯、乙苯等芳香族烃系溶剂及N,N-二甲基甲酰胺、二甲基亚砜、N,N-二甲基乙酰胺、N-甲基-2-吡咯烷酮、乙腈等非质子性极性有机溶剂,更优选为甲苯、二甲苯、N,N-二甲基甲酰胺、N-甲基-2-吡咯烷酮、甲苯与N,N-二甲基甲酰胺的混合溶剂、二甲苯与N,N-二甲基甲酰胺的混合溶剂或二甲苯与N-甲基-2-吡咯烷酮的混合溶剂。作为同时使用的试剂,可列举N,N’-二环己基碳二亚胺、1-乙基-3-(3-二甲基氨基丙基)碳二亚胺盐酸盐、五氧化二磷、硫酸、多聚磷酸、亚硫酰氯、三氯氧磷、乙二酰氯等,这些试剂优选使用0.2~5.0当量。另外,在作为同时使用的试剂使用氯化锌、氯化铜、氯化镁、氯化钴、氯化镍、氯化亚铁、氯化铝、硫酸铁、硫酸铝、三氟化硼、对甲苯磺酸等时,优选使用0.0001~1.0当量。反应在使用五氧化二磷、硫酸、多聚磷酸、亚硫酰氯、三氯氧磷、乙二酰氯、氯化锌、氯化铜、氯化镁、氯化钴、氯化镍、氯化亚铁、氯化铝、硫酸铁、硫酸铝、三氟化硼、对甲苯磺酸时,优选在碱不存在下进行,在使用N,N’-二环己基碳二亚胺、1-乙基-3-(3-二甲基氨基丙基)碳二亚胺盐酸盐时,优选在碱存在下进行。使用碱时作为优选的碱,可列举碳酸钠、碳酸钾、碳酸氢钾、三乙胺、吡啶、二甲基氨基吡啶等,更优选为三乙胺。酰基化剂的使用量优选为1.0~5.0当量,更优选为1.0~3.0当量。使用亚硫酰氯、三氯氧磷、乙二酰氯时,这些试剂使用0.2~5.0当量,反应温度优选为-30℃~80℃的范围,更优选为-10℃~40℃。使用五氧化二磷、硫酸、多聚磷酸时,这些试剂使用0.2~5.0当量,反应温度优选为-30℃~200℃的范围,更优选为-10℃~160℃。使用N,N’-二环己基碳二亚胺、1-乙基-3-(3-二甲基氨基丙基)碳二亚胺盐酸盐时,优选这些试剂使用0.2~5.0当量,反应温度优选为-30℃~80℃的范围,更优选为-10℃~40℃,以三乙胺作为碱使用0.2~5.0当量的条件。使用氯化锌、氯化铜、氯化镁、氯化钴、氯化镍、氯化亚铁、氯化铝、硫酸铁、硫酸铝、三氟化硼、对甲苯磺酸时,这些试剂使用0.0001~1.0当量,反应温度优选为20℃~200℃的范围,更优选为80℃~160℃。反应时间优选为0.1小时~7天的范围,更优选为0.5小时~2天的范围。
特别优选的条件是,作为酰基化剂使用三氟乙酸,作为溶剂使用甲苯、N,N-二甲基甲酰胺、二甲苯、N-甲基-2-吡咯烷酮、N,N-二甲基乙酰胺、N,N-二甲基甲酰胺与甲苯的混合溶剂、二甲苯与N,N-二甲基甲酰胺的混合溶剂、二甲苯与N-甲基-2-吡咯烷酮的混合溶剂或二甲苯与N,N-二甲基乙酰胺的混合溶剂,酰基化剂的使用量为1.0~3.0当量。使用亚硫酰氯、三氯氧磷、乙二酰氯时的特别优选的条件是,这些试剂使用0.3~3.0当量,在碱不存在下,反应温度为-10℃~40℃,反应时间为0.5小时~1天的条件。使用五氧化二磷、硫酸、多聚磷酸时的特别优选的条件是,这些试剂使用0.2~2.0当量,反应温度为-10℃~160℃、反应时间为0.5小时~1天的条件。使用N,N’-二环己基碳二亚胺、1-乙基-3-(3-二甲基氨基丙基)碳二亚胺盐酸盐时的特别优选的条件是,这些试剂使用0.5~3.0当量,反应温度为-10℃~40℃,以三乙胺作为碱使用0.5~3.0当量,反应时间为0.5~1天的条件。使用氯化锌、氯化铜、氯化镁、氯化钴、氯化镍、氯化亚铁、氯化铝、硫酸铁、硫酸铝、三氟化硼、对甲苯磺酸时的特别优选的条件是,这些试剂使用0.0001~0.5当量,在碱不存在下,反应温度为80℃~160℃,反应时间为2小时~2天的条件。
(5)在R2表示卤原子时,具体地在使用三氟乙酰氯、三氟乙酰溴,优选为三氟乙酰氯时,作为优选的溶剂,可列举甲苯、二甲苯、乙苯等芳香族烃系溶剂、二氯甲烷、氯仿等卤素系溶剂、及N,N-二甲基甲酰胺、二甲基亚砜、N,N-二甲基乙酰胺、N-甲基-2-吡咯烷酮、乙腈等非质子性极性有机溶剂,更优选为甲苯、N,N-二甲基甲酰胺、N-甲基-2-吡咯烷酮、或它们的混合溶剂。反应优选在碱不存在下进行,但在使用碱的情况下,作为优选的碱,可列举碳酸钠、碳酸钾、碳酸氢钾、三乙胺、吡啶等,更优选为碳酸钾。酰基化剂的使用量优选为1.0~5.0当量,更优选为1.0~3.0当量。使用碱时的碱使用量优选为1.0~5.0当量,更优选为1.0~3.0当量。反应温度优选为-80℃~40℃的范围,更优选为-30℃~30℃。反应时间优选为0.1小时~7天的范围,更优选为0.5小时~8小时的范围。
另外,R2表示氯原子时,还可以使用在与式(Aa)所表示的化合物反应的体系外同时使用三氟乙酸与亚硫酰氯、三氯氧磷、乙二酰氯等而预先生成的R1COCl。
特别优选的条件是,作为酰基化剂使用三氟乙酰氯,作为溶剂使用甲苯、N,N-二甲基甲酰胺、N-甲基-2-吡咯烷酮或它们的混合溶剂,酰基化剂的使用量为1.0~3.0当量,反应温度为-30℃~30℃,反应时间为0.5小时~8小时的条件。特别优选在碱不存在下,或在使用碱的情况下使用碳酸钾、其当量数使用1.0~3.0当量的条件。
式(Ba)所表示的化合物可以通过专利文献3等所记载的方法获得。即,作为从式(Aa)所表示的化合物制造式(Ba)所表示的化合物的方法,可以在无溶剂条件下或在不影响反应的溶剂中,使式(Aa)所表示的化合物在碱存在下或不存在下与式(Ca)所表示的化合物(X、R3、R4表示与上述所定义的相同的含义)所表示的化合物反应,从而得到。
作为可用的溶剂,可列举乙醚、异丙醚、四氢呋喃、二烷等醚系溶剂、N,N-二甲基甲酰胺、二甲基亚砜、N,N-二甲基乙酰胺、乙腈、N-甲基-2-吡咯烷酮、N-甲基-2-哌嗪酮、N,N-二甲基-2-咪唑烷酮等非质子性极性有机溶剂、二氯甲烷、氯仿等卤素系溶剂、甲苯、二甲苯、乙苯等芳香族烃系溶剂、及它们的混合溶剂,优选列举非质子性极性有机溶剂。更优选为N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、甲苯。
即使不使用碱也可实施反应,但作为使用碱时的可用的碱,可列举碳酸钠、碳酸钾、碳酸氢钠、碳酸氢钾、氢氧化钠、氢氧化钾、氢氧化镁、氢氧化钙、氢氧化锂、氢氧化钡等无机碱、1,8-二氮杂二环[5.4.0]十一碳-7-烯、1,5-二氮杂二环[4.3.0]壬-5-烯、三乙胺、二异丙基乙基胺、吡啶、二甲基吡啶、N,N-二甲基苯胺、N,N-二乙基苯胺、二甲基氨基吡啶等有机碱,优选列举碳酸钾、三乙胺、吡啶等,更优选列举三乙胺、碳酸钾。
使用碱时的碱使用量优选相对于式(Aa)所表示的化合物为1.0~3.0当量,更优选为1.1~2.5当量。反应温度优选为-20℃~150℃的范围,更优选为-10℃~100℃。
反应时间优选为0.1小时~7天的范围,更优选为1小时~2天。
另外,作为得到式(Ba)所表示的化合物其它方法,可列举将所述式(Ib)所表示的化合物水解来制造式(Ba)所表示的化合物的方法(式中,R1、R3、X表示与所述的定义相同的含义)。
作为可用的溶剂,可列举乙醚、异丙醚、四氢呋喃、二烷等醚系溶剂、N,N-二甲基甲酰胺、二甲基亚砜、N,N-二甲基乙酰胺、乙腈、N-甲基-2-吡咯烷酮、N-甲基-2-哌嗪酮、N,N-二甲基-2-咪唑烷酮等非质子性极性有机溶剂、二氯甲烷、氯仿等卤素系溶剂、甲苯、二甲苯、乙苯等芳香族烃系溶剂、甲醇、乙醇等醇系溶剂、水、及它们的混合溶剂,优选为芳香族烃系溶剂、或非质子性极性有机溶剂、或醇系溶剂与水的混合溶剂,更优选为N,N-二甲基甲酰胺与水、甲醇与水、或甲苯与水的混合溶剂。作为酸,可使用盐酸、硫酸、磷酸、硝酸等无机酸,作为碱,可使用碳酸钠、碳酸钾、碳酸氢钠、碳酸氢钾、氢氧化钠、氢氧化镁、氢氧化钙、氢氧化锂、氢氧化钡等无机碱。反应温度优选为-20℃~150℃的范围,更优选为70℃~100℃。反应时间优选为0.1小时~7天的范围,更优选为1小时~8小时。
从式(Aa)所表示的化合物经由式(Ba)所表示的化合物合成式(Ia)所表示的化合物时,可以不分离式(Ba)所表示的化合物地进行之后的工序,得到式(Ia)所表示的化合物。
从式(A)或式(Aa)所表示的化合物合成式(I)或式(Ia)所表示的化合物时,可以使酰基化剂、溶剂、Ar-CH2-R4、碱一次反应,而得到式(I)或式(Ia)所表示的化合物。
作为从式(A)或式(Aa)所表示的化合物使酰基化剂、溶剂、Ar-CH2-R4、碱一次反应而得到式(I)或式(Ia)所表示的化合物时的优选例,是将式(A)或式(Aa)所表示的化合物,使用甲苯、二甲苯、乙苯等芳香族烃系溶剂、N,N-二甲基甲酰胺、二甲基亚砜、N,N-二甲基乙酰胺、乙腈、N-甲基-2-吡咯烷酮等非质子性溶剂或其混合溶剂,使用相对于式(A)或(Aa)所表示的化合物为1.0~5.0当量的R2表示可被卤原子取代的C1-6烷氧基的酰基化剂、具体为三氟乙酸乙酯、三氟乙酸甲酯、三氟乙酸丙酯等,作为碱使用相对于式(A)或(Aa)所表示的化合物为1.0~10.0当量的碳酸钠、碳酸钾、碳酸氢钠、碳酸氢钾、氢氧化钠、氢氧化镁、氢氧化钙、氢氧化锂、氢氧化钡等无机碱、1,8-二氮杂二环[5.4.0]十一碳-7-烯、1,5-二氮杂二环[4.3.0]壬-5-烯、三乙胺、二异丙基乙基胺、吡啶、甲基吡啶、二甲基氨基吡啶等有机碱、乙醇钠、甲醇钠、叔丁醇钾等醇化物,加入相对于式(A)或(Aa)所表示的化合物为0.8~1.5当量的Ar-CH2-R4,在20℃~100℃下反应2小时~3天,得到式(I)或式(Ia)所表示的化合物的方法。
特别优选为使用甲苯、N,N-二甲基甲酰胺、或甲苯与N,N-二甲基甲酰胺的混合溶剂作为溶剂,使用三氟乙酸乙酯作为酰基化剂,Ar-CH2-R4的R4为氯原子,作为碱使用碳酸钾,相对于式(I)或式(Ia)所表示的化合物,酰基化剂的当量数优选为1.0~5.0当量,更优选为1.5~5.0当量,Ar-CH2-R4的当量数为0.8~1.5当量,碱的当量数为1.0~5.0当量,反应温度为40℃~80℃,反应时间为4小时~2天的条件。
从粗生成物纯化分离式(I)所表示的化合物及式(Ia)所表示的化合物的方法
式(I)所表示的化合物及式(Ia)所表示的化合物可以通过单独或组合通常使用的结晶化法、溶剂提取法、柱层析等进行纯化分离。溶剂提取法所使用的溶剂只要是不与水混合的溶剂就不特别选择,具体可列举乙酸乙酯、乙酸丁酯、甲苯、乙苯、乙醚、异丙醚、二氯甲烷、氯仿等。结晶化法中使用的溶剂,可列举水、己烷、甲苯、丙酮、N,N-二甲基甲酰胺、甲醇、2-丙醇、二氯甲烷、氯仿、乙酸乙酯、乙醚、二甲苯、N-甲基-2-吡咯烷酮、N,N-二甲基乙酰胺等及它们的混合溶剂。
式(I)所表示的化合物及式(Ia)所表示的化合物的优选的纯化分离方法为结晶化法,作为结晶化溶剂优选单独或组合使用丙酮、甲苯、水、N,N-二甲基甲酰胺、甲醇、二甲苯、N-甲基-2-吡咯烷酮、N,N-二甲基乙酰胺,更优选为水、甲醇、N,N-二甲基甲酰胺、N-甲基-2-吡咯烷酮、N,N-二甲基乙酰胺的组合。
实施例
本发明的具体例在以下显示,但是本发明并不限于这些。
合成例1:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺(化合物1)的合成
(1)将25g(270mmol)2-氨基吡啶溶解于200ml无水二氯甲烷中,加入41ml(30g,300mmol)三乙胺并冷却至0℃。在其中经过15分钟滴加38ml(57g,270mmol)三氟乙酸酐,在室温搅拌2小时。反应结束后,将反应液注入约100ml的冰水中,搅拌10分钟。移到分液漏斗进行分液,将有机层以150ml水洗涤2次,并以150ml的1%HCL水溶液洗涤2次后,以无水硫酸镁进行干燥,减压浓缩而得到36g的2,2,2-三氟-N-(吡啶-2(1H)-亚基)乙酰胺(收率71%)。
1H-NMR(CDCl3,δ,ppm):7.20(1H,m),7.83(1H,m),8.20(1H,d),8.35(1H,d),10.07(1H,brs)
13C-NMR(CDCl3,δ,ppm):115.3,115.5(q),121.6,139.1,147.9,149.5,155.3(q)MS:m/z=191(M+H)。
(2)将20g(126mmol)2-氯-5-氯甲基吡啶溶解于200ml无水乙腈,加入24g(126mmol)上述的方法所得到的2,2,2-三氟-N-(吡啶-2(1H)-亚基)乙酰胺、21g(151mmol)碳酸钾,加热回流6小时后,在室温搅拌10小时。反应结束后,过滤反应液,减压浓缩滤液。在其中加入乙醚而结晶化,滤取所得到的晶体,以乙醚与水充分洗涤。所得到的晶体于60℃下减压干燥1小时,得到目的物。产量26g(收率66%)。
1H-NMR(CDCl3,δ,ppm):5.57(2H,s),6.92(1H,td),7.31(1H,d),7.80(1H,td),7.87(1H,dd),7.99(1H,dd),8.48(2H,m)
13C-NMR(CDCl3,δ,ppm):53.8,115.5,117.2(q),122.1,124.7,130.0,139.2,140.0,142.5,149.7,151.8,158.9,163.5(q)
MS:m/z=316(M+H)。
(3)粉末X射线晶体分析
在粉末X射线衍射中,用以下的条件进行测定。
装置名:RINT-2200(株式会社リガク)
X射线:Cu-Kα(40kV、20mA)
扫描范围:4~40°取样宽度:0.02°扫描速度:1°/分钟
结果如下(图1)。
衍射角(2θ)8.7°、14.2°、17.5°、18.3°、19.8°、22.4°、30.9°、35.3°。
(4)差示扫描量热分析(DSC)
在差示扫描量热分析中,在以下的条件下测定。
装置名:DSC-60
样品盒:铝
温度范围:50℃~250℃(升温:10℃/分钟)
结果在图2中显示。
(5)另外,通过以下的(i)~(iv)所记载的方法(第二~五的制法)进行再晶体而得到同等的晶体。对这些晶体,以与上述同样的测定条件,进行粉末X射线晶体分析及差示扫描量热分析。
(i)第二制法
在化合物1(700mg)中加入约25ml己烷、约25ml乙酸乙酯,在热水浴中加热至65℃使其完全溶解。将其缓缓恢复到室温并放置一夜。滤出所析出的晶体,以少量的己烷:乙酸乙酯=95:5的溶液洗涤晶体。将其放入干燥器在减压下干燥2小时,得到白色晶体349mg。
粉末X射线晶体分析的结果如下(图3)。
衍射角(2θ)8.5°、14.0°、17.3°、18.1°、19.6°、22.2°、30.8°、35.2°
差示扫描量热分析的结果在图4中显示。
(ii)第三制法
在化合物1(1.0g)中加入28ml 2-丙醇,在热水浴中加热至65℃使其完全溶解。使其缓缓恢复到室温并放置一夜。滤出所析出的晶体,以少量的2-丙醇洗涤后,置入干燥器中减压下干燥2小时,得到白色晶体695mg。
差示扫描量热分析的结果在图5中显示。
(iii)第四制法
在化合物1(700mg)中加入约30ml甲苯,在热水浴中加热至65℃使其完全溶解。将其缓缓恢复到室温并放置一夜。滤出所析出的晶体,以少量的甲苯洗涤后,置入干燥器中减压下干燥2小时,得到白色晶体440mg。
粉末X射线晶体分析的结果如下(图6)。
衍射角(2θ)8.6°、14.2°、17.5°、18.3°、19.7°、22.3°、30.9°、35.3°
差示扫描量热分析的结果在图7中显示。
(iv)第五制法
在化合物1(50mg)中加入甲醇约2ml、水约2ml,在热水浴中加热至65℃使其溶解。将其恢复到室温并放置一夜。滤出所析出的晶体,得到白色晶体16mg。
差示扫描量热分析的结果在图8中显示。
表1显示依照合成例1的方法所制造的式(I)所表示的有害生物防除剂的具体化合物例及其物性值。
表1
合成例2:2,2,2-三氟-N-(吡啶-2(1H)-亚基)乙酰胺的合成
将1.0g(10.6mmol)2-氨基吡啶溶解于10ml乙酸乙酯中,加入1.78ml(12.7mmol)三乙胺,冰冷下添加1.62ml(11.7mmol)三氟乙酸酐。然后在室温搅拌2小时后,加入10ml乙酸乙酯与10ml水并搅拌后进行分液。将乙酸乙酯层进一步以10ml水洗涤二次后,以无水硫酸镁干燥,减压浓缩,得到2,2,2-三氟-N-(吡啶-2(1H)-亚基)乙酰胺1.56g(77.2%)。
合成例3:2,2,2-三氟-N-(吡啶-2(1H)-亚基)乙酰胺的合成
将4.7g(50mmol)2-氨基吡啶溶解于25ml N,N-二甲基甲酰胺中,添加35.5g(250mmol)三氟乙酸乙酯。然后在55~60℃搅拌15小时后,加入100ml乙酸乙酯和100ml水并搅拌后进行分液。将乙酸乙酯层进一步以100ml水及100mL食盐水洗涤后,以无水硫酸镁干燥,减压浓缩,得到2,2,2-三氟-N-(吡啶-2(1H)-亚基)乙酰胺9.05g(95.6%)。
1H-NMR(CDCl3,δ,ppm):7.20(1H,ddd),7.83(1H,td),8.20(1H,d),8.35(1H,d),10.07(1H,brs)。
合成例4:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的合成
将50.0g(0.53mol)2-氨基吡啶溶解于400ml甲苯后,在5℃冷却下,将88.6ml(0.64mol)三氟乙酸酐以30分钟滴加。滴加后在室温搅拌30分钟,减压下将20ml甲苯蒸馏。在反应液中加入250ml二甲基甲酰胺,冰冷下缓缓加入88.2(0.64mol)g粉末碳酸钾。然后,加入89.2g(0.557mol)2-氯-5-氯甲基吡啶,在减压(50-60hPa)下,在40-45℃将甲苯慢慢蒸馏,加热1小时。以60-70℃、35hPa进一步加热蒸馏2.5小时后,加入5.0g(0.036mol)粉末碳酸钾,以50-60℃、35hPa进一步1小时以除去水。将反应液添加在2L 50℃的水中,添加结束后,搅拌30分钟。然后,过滤,以200ml、接着以500ml水进行浆洗涤。过滤后,以甲苯100ml进行压洗,进一步以400ml甲苯进行浆洗涤。将所得到的晶体以60℃、真空泵减压下进行干燥一夜,得到作为目的的N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺147.78g(88.1%)。取8.21g的所得到的标题化合物,溶解于100mL丙酮,在其中加入300mL水。在室温搅拌并滤取所析出的晶体,将所得到的晶体以60℃、真空泵减压下干燥一夜,得到7.28g。所得到的晶体的粉末X射线晶体分析的结果如下(图9)。
衍射角(2θ)8.8°、14.3°、17.6°、18.3°、19.9°、22.5°、31.0°、35.4°
合成例5:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的合成
将50.0g(0.53mol)2-氨基吡啶溶解于250ml甲苯后,在5℃冷却下,将88.6ml(0.64mol)三氟乙酸酐以30分钟滴加。滴加后在室温搅拌30分钟,减压下蒸馏20ml甲苯。在反应液中加入250ml二甲基甲酰胺,在冰冷下,缓缓加入88.2g(0.64mol)粉末碳酸钾。然后,加入87.0g(0.54mol)2-氯-5-氯甲基吡啶,在减压(50-60hPa)下、以50-60℃缓缓蒸馏甲苯,进一步在35hPa下加热。1小时后,加入5.0g(0.036mol)粉末碳酸钾,以50-60℃、35hPa除去水。4小时后,将反应液添加在1.1L的50℃的水中,反应容器用150ml甲醇洗涤,并补加添加。添加结束后,在50℃加热10分钟,慢慢冷却,在15-20℃搅拌30分钟后,过滤,以150ml水、接着以150ml甲苯洗涤。浆所得到的晶体以60℃、真空泵减压下干燥11小时,得到作为目的的N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺147.32g(87.8%)。
合成例6:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的合成
将1.0g(10.6mmol)2-氨基吡啶溶解于10ml甲苯中,在5℃冷却后,加入1.18ml(15.9mmol)三氟乙酸,然后再加入0.99ml(10.6mmol)三氯氧磷,室温下搅拌6.5小时。在反应液中加入5.0ml二甲基甲酰胺与5.87g(42.5mmol)粉末碳酸钾及1.72g(10.6mmol)2-氯-5-氯甲基吡啶,在减压下(60-35hPa)、以50-60℃蒸馏。2.5小时后,将反应液添加到100ml的水中,过滤晶体,以30ml水和15ml甲苯洗涤。所得到的晶体减压下60℃干燥,得到作为目的的化合物N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺2.09g(62.3%)。
合成例7:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的合成
将10.0g(0.106mol)2-氨基吡啶溶解于100ml甲苯中,在5℃冷却后,加入11.8ml(0.159mol)三氟乙酸,然后再加入9.9ml(0.106mol)氯氧化磷,室温下搅拌一夜,在减压下,将20ml甲苯馏去。在冰冷下,在反应液中加入50ml二甲基甲酰胺与35.28g(0.256mol)粉末碳酸钾、及17.22g(0.106mol)2-氯-5-氯甲基吡啶,在减压下(60-35hPa)、以50-60℃蒸馏。2小时后,进一步加入25ml二甲基甲酰胺、20ml甲苯与7.35g(0.053mol)粉末碳酸钾,在减压下(60-35hPa)、以50-60℃蒸馏2小时。在反应液中加入60ml甲醇与50ml水,在一并洗涤容器的同时将反应液添加到300ml的水中,30分钟后,过滤晶体,以70ml水与40ml甲苯洗涤。所得到的晶体在减压下60℃干燥,得到作为目的的化合物25.75g(76.9%)。
合成例8:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的合成
将10.0g(0.106mol)2-氨基吡啶溶解于100ml甲苯,在5℃冷却后,加入11.8ml(0.159mol)三氟乙酸,然后再加入7.7ml(0.106mol)亚硫酰氯,在室温下搅拌一夜,减压下将20ml甲苯馏去。在冰冷下,在反应液中加入50ml二甲基甲酰胺、35.28g(0.256mol)粉末碳酸钾与17.22g(0.106mol)2-氯-5-氯甲基吡啶,在减压下(36hPa)、以50-60℃蒸馏一小时。在反应液中加入60ml甲醇与50ml水,在一并洗涤容器的同时将反应液添加到300ml的水中,30分钟后,过滤晶体,以70ml水与40ml甲苯洗涤。所得到的晶体在减压下60℃干燥,得到作为目的的化合物22.31g(66.6%)。
合成例9:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的合成
将5.0g(0.053mol)2-氨基吡啶溶解于50ml甲苯后,在5℃冷却下,将8.86ml(0.064mol)三氟乙酸酐以10分钟滴加。滴加后于室温搅拌30分钟,在减压下蒸馏10ml甲苯。在反应液中加入25ml二甲基甲酰胺,冰冷下,缓缓加入8.82g粉末碳酸钾。然后,加入11.78g(0.053mol)2-氯-5-甲磺酰氧基甲基吡啶,在减压(50-60hPa)下、以50-60℃慢慢蒸馏甲苯,进一步在35hPa进行加热。30分钟后,加入30ml二甲基甲酰胺、30ml甲苯与1.18g(0.0053mol)2-氯-5-甲磺酰氧基甲基吡啶,以50-60℃、55hPa进行减压蒸馏。4小时后,将反应液添加到250ml水中,将反应容器用30ml甲醇与20ml水洗涤并补加添加。添加结束后,于室温搅拌30分钟后,过滤,以50ml水接着再以40ml甲苯洗涤。将所得到的晶体以80℃、真空泵减压下干燥11小时,得到作为目的的N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺11.63g(69.4%)。
合成例10:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的合成
将10.0g(0.106mol)2-氨基吡啶溶解于100ml甲苯,在5℃冷却后,加入11.84ml(0.159mol)三氟乙酸,然后再加入5.94ml(0.064mol)三氯氧磷,于室温下搅拌一夜,减压下,馏去20ml甲苯。在冰冷下在反应液中加入50ml二甲基甲酰胺与22.03g(0.16mol)粉末碳酸钾及17.56g(0.108mol)2-氯-5-氯甲基吡啶,以减压下(60-35hPa)、以50-60℃蒸馏。1小时后,进一步加入20ml二甲基甲酰胺、20ml甲苯与4.41g(0.032mol)粉末碳酸钾,在减压下(60-35hPa)、以50-60℃蒸馏1.5小时。在反应液中加入30ml甲醇,然后添加到250ml的50℃的水中,然后再加入水50ml,在洗涤容器的同时添加,于室温冷却后搅拌30分钟,过滤晶体,以50ml水与30ml甲苯洗涤。所得到的晶体在减压下60℃进行干燥,得到作为目的的化合物23.69g(70.6%)。
合成例11:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的合成
将10.0g(0.106mol)2-氨基吡啶溶解于100ml甲苯,在5℃冷却后,加入11.8ml(0.159mol)三氟乙酸,然后再分次加入7.76ml(0.106mol)亚硫酰氯,于室温下搅拌一夜,减压下,馏去50ml甲苯。在反应液中加入50ml甲苯,冰冷下加入50ml二甲基甲酰胺、22.03g(0.16mol)粉末碳酸钾与17.56g(0.108mol)2-氯-5-氯甲基吡啶,在减压下(90-36hPa)、以60℃蒸馏1.5小时。在反应液中加入30ml甲醇与20ml水,与一并洗涤容器的同时将反应液添加到50℃的300mL的水中,于室温30分钟搅拌后,过滤晶体,以50ml水与30ml甲苯洗涤。所得到的晶体以减压下60℃干燥,得到作为目的的化合物21.45g(64.1%)。
合成例12:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的合成
将94g(1mol)2-氨基吡啶溶解于500mL二甲基甲酰胺,加入284g(2mol)三氟乙酸乙酯,于55-60℃搅拌24小时。在反应液中加入82.8g(0.6mol)粉末碳酸钾与153.9g(0.95mol)2-氯-5-氯甲基吡啶及300mL甲苯,在减压下(36hPa)、以50-60℃搅拌3小时。在反应液中添加200mL甲醇后,将反应液添加到2L的50℃的温水中,放冷至室温后,搅拌3小时。过滤晶体,以400mL水及450mL甲苯洗涤。所得到的晶体以减压下45℃干燥,得到作为目的的化合物228.9g(收率72.7%)。
合成例13:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的合成
将9.4g(0.1mol)2-氨基吡啶溶解于30mL二甲基甲酰胺及20mL甲苯的混合溶剂中,加入28.4g(0.2mol)三氟乙酸乙酯,于60-65℃搅拌8小时。在反应液中加入16.6g(0.12mol)粉末碳酸钾与16.2g(0.1mol)2-氯-5-氯甲基吡啶,以60-65℃搅拌15小时。在反应液中添加15mL甲醇后,在120mL的50℃的温水中添加反应液,放冷至室温后,搅拌2小时。过滤晶体,以50mL水及100mL甲苯洗涤。所得到的晶体以减压下45℃干燥,得到作为目的的化合物25.6g(收率81.2%)。
合成例14:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的合成
在13.68g(0.12mol)三氟乙酸中添加1.5mL二甲基甲酰胺,在65℃加温的同时加入14.28g(0.12mol)亚硫酰氯。将由此产生的三氟乙酰氯鼓泡至将9.4g(0.1mol)2-氨基吡啶溶解于50mL N-甲基吡咯烷酮并于-10℃冷却而得的溶液中,搅拌1小时。在该反应液中加入100mL甲苯、48.3g(0.35mol)粉末碳酸钾与16.52g(0.102mol)2-氯-5-氯甲基吡啶,减压下(36hPa)、以50-60℃蒸馏3小时。在反应液中加入20mL甲醇,在一并洗涤容器的同时添加到加温至50℃的300mL的水中。以室温搅拌1.5小时后,过滤晶体,以100mL水及150mL甲苯洗涤。将所得到的晶体以减压下45℃干燥,得到作为目的的化合物16.8g(收率53.3%)。
合成例15:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的合成
在18.24g(0.16mol)三氟乙酸中添加8.76g(0.12mol)二甲基甲酰胺,在65℃加温的同时加入12.26g(0.08mol)三氯氧磷。将由此产生的三氟乙酰氯鼓泡至将9.4g(0.1mol)2-氨基吡啶溶解于80mL N-甲基吡咯烷酮于-15℃冷却而得的溶液中,搅拌2小时。在-10℃冷却的同时,在该反应液中加入14.9g(0.22mol)粉末乙醇钠,进行中和。在反应液中加入13.8g(0.1mol)粉末碳酸钾、及16.2g(0.1mol)2-氯-5-氯甲基吡啶,减压下(36hPa)、以50-60℃蒸馏2小时。在反应液中加入20mL甲醇,在一并洗涤容器的同时添加到加温至50℃的400mL的水中。于室温搅拌30分钟后,过滤晶体,以100mL水及50mL甲苯洗涤。所得到的晶体以减压下45℃干燥,得到作为目的的化合物22.5g(收率71.4%)。
合成例16:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的合成
将3.00g(18.6mmol)2-氯-5-氯甲基吡啶溶解于20ml二甲基甲酰胺中,加入1.75g(18.6mmol)2-氨基吡啶在80℃搅拌8小时、在室温搅拌5小时。反应结束后,减压馏去二甲基甲酰胺,加入乙腈,结果析出固体,因此滤出,以乙腈彻底洗涤后进行干燥,得到1-[(6-氯吡啶-3-基)甲基]吡啶-2(1H)-亚胺盐酸盐2.07g(收率44%)。
1H-NMR(DMSO-d6,δ,ppm):5.65(2H,s),6.96(1H,t),7.23(1H,m),7.57(1H,d),7.80(1H,m),7.91(1H,m),8.28(1H,m),8.49(1H,d)。
将50mg(0.20mmol)以上述的方法得到的1-[(6-氯吡啶-3-基)甲基]吡啶-2(1H)-亚胺盐酸盐悬浮于5ml无水二氯甲烷中,在冰冷下依次加入122mg(1.00mmol)二甲基氨基吡啶、50mg(0.24mmol)三氟乙酸酐,在室温搅拌1小时。反应结束后,将反应液以二氯甲烷稀释,以1%盐酸洗涤后,以无水硫酸镁干燥。减压馏去二氯甲烷而得到目的物。产量42mg(收率67%)。
合成例17:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的合成
按照合成例16的方法合成后,将4.6g(0.02mol)中和而得到的1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚胺溶解于15mL N,N-二甲基甲酰胺中,加入5.7g(0.04mol)三氟乙酸乙酯。于56℃搅拌整夜后,加入60mL水,滤取所析出的晶体。将所得到的晶体在减压下45℃下干燥,得到作为目的的化合物5.85g(收率92.8%)。
合成例18:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的合成
按照合成例16的方法合成后,将2.2g(0.01mol)中和而得到的1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚胺溶解于6mL N,N-二甲基甲酰胺中,冰冷下,加入828mg(0.006mol)碳酸钾及2.52g(0.012mol)三氟乙酸酐。于室温搅拌1小时后,加入30mL水,滤取所析出的晶体。将所得到的晶体以20mL水洗涤后,减压下45℃下干燥,得到作为目的的化合物2.38g(收率75.6%)。
合成例19:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的合成
在4.56g(0.04mol)三氟乙酸中添加3mL N,N-二甲基甲酰胺,在60℃加温的同时加入3.12g(0.02mol)三氯氧磷。将由此产生的三氟乙酰氯,鼓泡至将4.38g(0.02mol)按照合成例16的方法合成后、中和而得到的1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚胺溶解于25mL N-甲基-2-吡咯烷酮而得的溶液中,于-10℃使其反应45分钟。加入125mL水,滤取所析出的晶体。将所得到的晶体在减压下45℃下干燥,得到作为目的的化合物2.58g(收率40.9%)。
合成例20:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的合成
按照合成例16的方法合成后,将4.38g(0.02mol)中和而得到的1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚胺溶解于3mL N,N-二甲基甲酰胺中,加入2.7g(0.024mol)三氟乙酸及2.8g(0.02mol)五氧化二磷。于120℃搅拌3小时后,回到室温,加入50mL水,滤取所析出的晶体。将所得到的晶体在减压下45℃下干燥,得到作为目的的化合物2.12g(收率33.7%)。
合成例21:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的合成
将9.4g(0.1mol)2-氨基吡啶溶解于50mL二甲基甲酰胺,加入28.8g(0.2mol)三氟乙酸乙酯与16.2g(0.1mol)2-氯-5-氯甲基吡啶、及13.8g(0.1mol)碳酸钾,于55-60℃搅拌20小时。在反应液中进一步加入1.38g(0.1mol)粉末碳酸钾与3.24(0.02mol)2-氯-5-氯甲基吡啶、及5.68g(0.04mol)三氟乙酸乙酯,于55-60℃搅拌6小时。在反应液中添加40mL甲醇然后,将反应液添加于300mL 50℃的温水中,放冷至室温后,搅拌1小时。过滤晶体,以100mL水及75mL甲苯洗涤。所得到的晶体在减压下45℃干燥,得到作为目的的化合物24.0g(收率76%)。
合成例22:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的合成
将9.4g(0.1mol)2-氨基吡啶溶解于30mL二甲基甲酰胺及20mL甲苯中,加入28.8g(0.2mol)三氟乙酸乙酯与16.2g(0.1mol)2-氯-5-氯甲基吡啶、及16.6g(0.12mol)碳酸钾,于60-65℃搅拌18小时。添加15mL甲醇于反应液中后,添加反应液于120mL 50℃的温水中,放冷至室温后,搅拌1小时。过滤晶体,以50mL水及100mL甲苯洗涤。所得到的晶体在减压下45℃下干燥,得到作为目的的化合物23.9g(收率75.9%)。
合成例23:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的合成
将4.7g(0.05mol)2-氨基吡啶溶解于25mL N,N-二甲基甲酰胺与10mL甲苯的混合溶剂中,加入35.5g(0.25mol)三氟乙酸乙酯与9.72g(0.06mol)2-氯-5-氯甲基吡啶、及8.28g(0.06mol)粉末碳酸钾,于65℃搅拌18小时。添加10mL甲醇于反应液后,添加反应液于150mL 50℃的温水中,放冷至室温后,搅拌1小时。过滤晶体,以50mL水及50mL甲苯洗涤。所得到的晶体在减压下45℃下干燥,得到作为目的的化合物13.78g(收率87.5%)。
合成例24:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的合成
将9.4g(0.1mol)2-氨基吡啶溶解于30mL二甲基甲酰胺及20mL甲苯中,加入14.2g(0.1mol)三氟乙酸乙酯于60-65℃搅拌7小时。接着加入16.2g(0.1mol)2-氯-5-氯甲基吡啶、及16.6g(0.12mol)碳酸钾,于60-65℃搅拌18小时。添加15mL甲醇于反应液然后,添加反应液于150mL 50℃的温水中,放冷至室温。过滤晶体,以50mL水及75mL甲苯洗涤。所得到的晶体在减压下60℃下干燥,得到作为目的的化合物20.6g(收率65.4%)。
合成例25:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的合成
将9.4g(0.1mol)2-氨基吡啶溶解于30mL二甲基甲酰胺及20mL甲苯中,加入7.1g(0.05mol)三氟乙酸乙酯于60-65℃搅拌7.5小时。经减压浓缩(90hPa、40℃)后,进行冰冷,加入20mL甲苯及10.5g(0.05mol)三氟乙酸酐,于室温搅拌1小时。接着,加入16.2g(0.1mol)2-氯-5-氯甲基吡啶、20mL二甲基甲酰胺、及16.6g(0.12mol)碳酸钾,于110hPa的减压下、于60-65℃搅拌4小时。经减压浓缩(90hPa、50℃)后,添加25mL甲醇于反应液中,将其加入250mL 50℃的温水中,放冷至室温并同时搅拌。过滤晶体,以90mL水及90mL甲苯洗涤。所得到的晶体在减压下60℃下干燥,得到作为目的的化合物19.8g(收率62.9%)。
合成例26:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的合成
将9.4g(0.1mol)2-氨基吡啶溶解于30mL二甲基甲酰胺及20mL甲苯中,加入21.3g(0.15mol)三氟乙酸乙酯于60-65℃搅拌7.5小时。经减压浓缩(90hPa、40℃)后,进行冰冷,加入20mL甲苯及10.5g(0.05mol)三氟乙酸酐,于室温搅拌1小时。接着,加入16.2g(0.1mol)2-氯-5-氯甲基吡啶、20mL二甲基甲酰胺、及16.6g(0.12mol)碳酸钾,于110hPa的减压下,于60-65℃搅拌4小时。经减压浓缩(90hPa、50℃)后,添加25mL甲醇于反应液中,将其加入250mL 50℃的温水中,放冷至室温同时搅拌。过滤晶体,以90mL水及90mL甲苯洗涤。所得到的晶体在减压下60℃下干燥,得到作为目的的化合物22.68g(收率72.0%)。
合成例27:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺的合成
将2.35g(0.025mol)2-氨基吡啶悬浮于40mL二甲苯中,加入2.85g(0.025mol)三氟乙酸及135mg氯化亚铁6水合物,在加入迪安-斯达克回流装置除去生成的水的同时,于150℃搅拌16小时。冷却至60℃后,加入4.05g(0.025mol)2-氯-5-氯甲基吡啶、16mL二甲基甲酰胺、及2.42g(0.0175mol)碳酸钾,于60-110hPa的减压下,于60-65℃搅拌3小时。添加10mL甲醇于反应液中,将其加入80mL的50℃的温水中,放冷至室温同时搅拌。过滤晶体,以20mL水及20mL甲苯洗涤。所得到的晶体在减压下60℃下干燥,得到作为目的的化合物6.32g(收率80.3%)。
合成例28:N-[1-((6-氯-5-氟吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺(化合物2)的合成
将4.00g(27.6mmol)2-氯-3-氟-5-甲基吡啶溶于80ml四氯化碳中,加入7.37g(41.4mmol)N-溴琥珀酰亚胺、20mg过氧化苯甲酰加热回流一夜。反应结束后,使反应液回到室温进行减压浓缩,用硅胶柱层析(己烷:乙酸乙酯=19:1)进行纯化得到5-(溴甲基)-2-氯-3-氟吡啶3.06g(收率51%)。
1H-NMR(CDCl3,δ,ppm):4.45(2H,s),7.54(1H,dd),8.23(1H,s)。
将50mg(0.22mmol)上述的方法所得到的5-(溴甲基)-2-氯-3-氟吡啶溶于5ml无水乙腈,依次加入42mg(0.22mmol)所述的方法所得到的2,2,2-三氟-N-(吡啶-2(1H)-亚基)乙酰胺、36mg(0.26mmol)碳酸钾,并加热回流7小时。反应结束后,使反应液回到室温过滤不溶物,减压浓缩滤液。在其中加入乙醚,结果析出固体,因而进行滤取,以乙醚洗涤后以干燥器减压干燥得到目的物。产量29mg(收率40%)。
1H-NMR(CDCl3,δ,ppm):5.54(2H,s),6.89(1H,td),7.76(1H,dd),7.80(1H,td),7.85(1H,d),8.29(1H,d),8.57(1H,d)
MS:m/z=334(M+H)。
合成例29:N-[1-((6-溴吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺(化合物3)的合成
将500mg(2.92mmol)2-溴-5-甲基吡啶溶解于15ml四氯化碳中,加入623mg(3.50mmol)N-溴琥珀酰亚胺、10mg过氧化苯甲酰并加热回流19小时。反应结束后,使反应液回到室温进行减压浓缩,以硅胶柱层析(己烷:乙酸乙酯=19:1)进行纯化得到2-溴-5-溴甲基吡啶143mg(收率20%)。
1H-NMR(CDCl3,δ,ppm):4.42(2H,s),7.47(1H,d),7.59(1H,dd),8.38(1H,d)。
将70mg(0.28mmol)以上述的方法所得到的2-溴-5-溴甲基吡啶溶于10ml无水乙腈中,依次加入54mg(0.28mmol)所述的方法所合成的2,2,2-三氟-N-(吡啶-2(1H)-亚基)乙酰胺、46mg(0.34mmol)碳酸钾,并加热回流6小时。反应结束后,使反应液回到室温过滤不溶物,减压浓缩滤液。在其中加入乙醚,结果析出固体,因而进行滤取,以乙醚洗涤后以干燥器进行减压干燥得到目的物。产量81mg(收率82%)。
1H-NMR(CDCl3,δ,ppm):5.52(2H,s),6.88(1H,t),7.48(1H,d),7.78(2H,m),7.84(1H,d),8.44(1H,d),8.53(1H,d)
MS:m/z=360(M+H)。
合成例30:2-氯-N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2-二氟乙酰胺(化合物4)的合成
将200mg(2.13mmol)2-氨基吡啶溶解于5ml二氯甲烷中,依次加入491mg(2.55mmol)EDC-HCl、311mg(2.55mmol)二甲基氨基吡啶、187μl(2.23mmol、290mg)氯二氟乙酸,并搅拌一夜。反应结束后,将反应液以二氯甲烷稀释,以水、1%盐酸洗涤后,以无水硫酸镁干燥得到2-氯-2,2-二氟-N-(吡啶-2(1H)-亚基)乙酰胺105mg(收率24%)。
1H-NMR(CDCl3,δ,ppm):7.19(1H,dd),7.82(1H,m),8.18(1H,d),8.36(1H,d),9.35(1H,brs)。
在68mg(0.33mmol)所述的方法所合成的2-氯-2,2-二氟-N-(吡啶-2(1H)-亚基)乙酰胺中,加入溶解于6ml无水乙腈的53mg(0.33mmol)2-氯-5-氯甲基吡啶,接着加入50mg(0.36mmol)碳酸钾加热回流1小时。反应结束后,使反应液回到室温然后进行减压浓缩。在其中加入乙醚,结果析出固体,因而进行滤取,进行干燥而得到目的物。产量49mg(收率45%)。
1H-NMR(CDCl3,δ,ppm):5.56(2H,s),6.92(1H,t),7.33(1H,d),7.82(1H,m),7.91(1H,dd),8.02(1H,d),8.45(1H,d),8.48(1H,d)
13C-NMR(CDCl3,δ,ppm):53.8,115.2,120.1(t),122.1,124.8,139.0,140.0,142.3,150.0,151.9,159.1,159.1,165.8(t)
MS:m/z=332(M+H)。
合成例31:2,2,2-三氯-N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]乙酰胺(化合物5)的合成
将70mg(0.27mmol)合成例16的方法所得到的1-[(6-氯吡啶-3-基)甲基]吡啶-2(1H)-亚胺盐酸盐悬浮于4ml无水二氯甲烷中,依次加入94μl(0.68mmol、68mg)三乙胺、33μg(0.27mmol、49mg)三氯乙酰氯,室温搅拌1小时。反应结束后,加入水使反应停止,以二氯甲烷与水进行分液。将有机层以水洗涤1次、以1%盐酸洗涤2次后,以无水硫酸镁干燥、减压浓缩。在其中加入乙醚,结果析出固体,因而进行滤取,干燥得到目的物。产量61mg(收率62%)。
1H-NMR(CDCl3,δ,ppm):5.59(2H,s),6.86(1H,t),7.32(1H,d),7.78(1H,td),7.91(2H,m),8.43(1H,d),8.50(1H,d)
MS:m/z=364(M+H)。
合成例32:N-[1-((2-氯嘧啶-5-基)甲基)吡啶-2(1H)-亚基]-2,2,2-三氟乙酰胺(化合物6)的合成
将1.04g(8.13mmol)2-氯-5-甲基嘧啶溶于30ml四氯化碳中,加入1.73g(9.75mmol)N-溴琥珀酰亚胺、20mg过氧化苯甲酰,加热回流6小时。反应结束后,使反应液回到室温进行减压浓缩,以硅胶柱层析(己烷:乙酸乙酯=3:1)进行纯化,得到5-溴甲基-2-氯嘧啶641mg(收率38%)。
1H-NMR(CDCl3,δ,ppm):4.42(2H,s),8.66(2H,s)。
将104mg(0.50mmol)上述的方法所得到的5-溴甲基-2-氯嘧啶溶解于6ml无水乙腈中,加入96mg(0.50mmol)所述的方法所得到的2,2,2-三氟-N-(吡啶-2(1H)-亚基)乙酰胺、76mg(0.55mmol)碳酸钾,加热回流1小时。反应结束后,使反应液回到室温过滤不溶物并除去,减压浓缩滤液。在其中加入乙醚,结果析出固体,因而进行滤取,以乙醚洗涤后放入干燥器中进行减压干燥,得到目的物。产量92mg(收率58%)。
1H-NMR(CDCl3,δ,ppm):5.54(2H,s),6.98(1H,m),7.87(1H,m),8.18(1H,m),8.48(1H,m),8.83(2H,m)
13C-NMR(CDCl3,δ,ppm):0.0,115.6,117.1(q),122.1,127.5,139.2,142.9,158.8,160.3(2C),161.4,163.8(q)
MS:m/z=317(M+H)。
合成例33:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2,3,3,3-五氟丙酰胺(化合物7)的合成
将300mg(3.19mmol)2-氨基吡啶溶解于15ml无水二氯甲烷中,依次加入919mg(4.78mmol)EDC-HCl、583mg(4.78mmol)DMAP、397μl(628mg、3.83mmol)五氟丙酸,于室温搅拌一夜。反应结束后,将反应液以二氯甲烷稀释,以水洗涤1次,以1%盐酸洗涤2次后,以无水硫酸镁干燥,减压浓缩,得到2,2,3,3,3-五氟-N-(吡啶-2(1H)-亚基)丙酰胺85mg(收率11%)。
在77mg(0.32mmol)上述的方法所得到的2,2,3,3,3-五氟-N-(吡啶-2(1H)-亚基)丙酰胺中,加入溶解于8ml无水乙腈的52mg(0.32mmol)2-氯-5-氯甲基吡啶、49mg(0.35mmol)碳酸钾,加热回流11小时。反应结束后,使反应液回到室温过滤不溶物,减压浓缩滤液。以硅胶柱层析(己烷:乙酸乙酯=1:3)进行纯化得到目的物。产量12mg(收率10%)。
1H-NMR(CDCl3,δ,ppm):5.56(2H,s),6.90(1H,td),7.32(1H,d),7.79(2H,m),7.84(1H,d),8.43(1H,d),8.56(1H,d)
MS:m/z=366(M+H)。
合成例34:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亚基]-2,2-二氟乙酰胺(化合物8)的合成
将400mg(4.26mmol)2-氨基吡啶溶解于10ml无水二氯甲烷中,加入322μl(490mg、5.11mmol)二氟乙酸、982mg(5.10mmol)EDC-HCl、622mg(5.11mmol)DMAP,于室温搅拌61小时。反应结束后,将反应液以二氯甲烷稀释,以水洗涤1次、1%HCl水溶液洗涤2次后,以无水硫酸镁干燥,减压浓缩得到2,2-二氟-N-(吡啶-2(1H)-亚基)乙酰胺102mg(收率14%)。
1H-NMR(CDCl3,δ,ppm):6.03(1H,t),7.15(1H,m),7.78(1H,td),8.20(1H,d),8.34(1H,dd),8.72(1H,brs)。
将100mg(0.58mmol)所述的方法所得到的2,2-二氟-N-(吡啶-2(1H)-亚基)乙酰胺溶解于10ml无水乙腈中,加入溶解于5ml无水乙腈的94mg(0.58mmol)2-氯-5-氯甲基吡啶,接着加入84mg(0.63mmol)碳酸钾,加热回流140分钟。反应结束后,使反应液回到室温,过滤除去不溶物,进行减压浓缩。在其中加入醚而析出固体,因而进行滤取,充分干燥得到目的物。产量63mg(收率37%)。
1H-NMR(CDCl3,δ,ppm):5.52(2H,s),5.90(1H,t),6.79(1H,td),7.33(1H,d),7.71(1H,m),7.77(1H,dd),7.85(1H,dd),8.45(1H,d),8.50(1H,d)
13C-NMR(DMSO-d6,δ,ppm):53.0,111.0(t),115.2,120.7,124.7,131.7,140.6,141.6,143.2,150.4,150.9,158.3,169.4(t)
MS:m/z=298(M+H)。
试验例1
小菜蛾防除试验
从槽栽培的卷心菜切下直径5.0cm的叶盘,在其上散布调制成50%丙酮水(加用0.05%Tween20)的500ppm的式(I)所表示的化合物的药液。风干后,在其中放饲2龄幼虫。然后,将其放置于25℃的恒温室(16小时光照期-8小时黑暗期)。放饲3日后观察虫的生死,依据下式算出死虫率。试验通过2连制进行。
死虫率(%)={死亡虫数/(生存虫数+死亡虫数)}×100。
试验例2
棉蚜防除试验
从槽栽培的黄瓜切除直径2.0cm的叶盘,在其上散布调制成50%丙酮水(加用0.05%Tween20)的500ppm的式(I)所表示的化合物的药液。风干后,在其中放饲1龄幼虫。然后,将其放置于25℃的恒温室(16小时光照期-8小时黑暗期)。放饲3日后观察虫的生死,依据下式算出死虫率。试验通过2连制进行。
死虫率(%)={死亡虫数/(生存虫数+死亡虫数)}×100。
试验例3
灰飞虱防除试验
将播种48小时后的小麦苗根部,用200μL的调制成10%丙酮水的100ppm的式(I)所表示的化合物的药液处理。72小时由根部吸收药剂,然后,在其中放饲灰飞虱的2龄幼虫各10只。然后,将其放置于25℃的恒温室(16小时光照期-8小时黑暗期)。放饲4日后观察虫的生死,依据下式算出死虫率。试验通过2连制进行。
死虫率(%)={死亡虫数/(生存虫数+死亡虫数)}×100。
作为试验例1~3的结果,表2显示式(I)所表示的有害生物防除剂的具体的生物活性(死虫率(%))。
表2
<对药剂低灵敏性害虫的效果>
参考例 褐飞虱防除试验
对槽栽培的稻苗,将调制成10%丙酮水的特定浓度的本发明化合物的药液进行土壤灌溉处理。处理3日后,于稻苗上放饲灵敏性、或药剂低灵敏性的褐飞虱2龄幼虫各10只。然后,将其放置于25℃的恒温室(16小时光照期-8小时黑暗期)。放饲3日后观察虫的生死,依据下式算出死虫率。试验通过2连制进行。
死虫率(%)={死亡虫数/(生存虫数+死亡虫数)}×100。
还有,试验害虫使用长是件室内累代饲育的虫(敏感性系统)、(I)2007年在熊本县内、(II)2005年在福冈县内分别采集后,在室内累代饲育的虫(野外采集系统)。
其结果显示,化合物1对任一系统在0.05mg/苗的处理中死虫率为100%,在0.005mg/苗的处理中死虫率为90%以上。另外,化合物2在0.01mg/苗的处理中,死虫率为(敏感性系统)72%,(II)70%,化合物19在0.01mg/苗的处理中,死虫率为(敏感性系统)100%、(II)93%。另一方面,吡虫啉在0.05mg/苗的处理中死虫率为(敏感性系统)100%、(I)40%、(II)60%。
其结果显示,化合物1对吡虫啉低灵敏性的褐飞虱,具有高的杀虫活性。
参考例 灰飞虱防除试验
对槽栽培的稻苗,将调制成10%丙酮水的特定浓度的本发明化合物的药液进行土壤灌溉处理。处理3日后,于稻苗上放饲灵敏性、或药剂低灵敏性的褐飞虱2龄幼虫各10只。然后,将其放置于25℃的恒温室(16小时光照期-8小时黑暗期)。放饲3日后观察虫的生死,依据下式算出死虫率。试验通过2连制进行。
死虫率(%)={死亡虫数/(生存虫数+死亡虫数)}×100。
此外,试验害虫使用长期间在室内累代饲育的虫(敏感性系统)、2006年在熊本县内采集后,在室内累代饲育的虫(野外采集系统)。
其结果显示,化合物1对任一系统在0.01mg/苗的处理中死虫率为100%,在0.005mg/苗的处理中死虫率为90%以上。另外,化合物3在0.01mg/苗的处理中死虫率为(敏感性系统)100%、(野外采集系统)90%。另一方面,吡虫啉在0.01mg/苗的处理中死虫率为(敏感性系统)100%、(野外采集系统)50%。另外,氟虫腈在0.01mg/苗的处理中死虫率为(敏感性系统)100%、(野外采集系统)70%。
该结果显示,化合物1、3对吡虫啉、及氟虫腈低灵敏性的灰飞虱具有高的杀虫活性。
参考例 化合物1及吡虫啉的使用家蝇粗酶提取液的体外代谢试验
如Pest Management Science(2003),59(3),347-352、及Journal of Pesticide Science(2004),29(2),110-116所记载,已知吡虫啉因受到氧化代谢而失活,认为是获得抵抗性的机制之一。为了确认对获得了这样的抵抗性的害虫的效果而进行以下的实验。
对家蝇成虫(0.645g),添加10mL的磷酸钾缓冲液(pH7.4,含有1mM EDTA),用Physcotron均质机(日音医理科器械制作所)充分磨碎。然后,以10,000g、15分钟的条件,将磨碎物进行离心分离。将所得到的上清液进一步以100,000g、60分钟的条件进行离心分离,得到沈淀物。将该沈淀物溶解于1mL的磷酸钾缓冲液中,以此作为粗酶液使用。酶提取作业全部在冰上或4℃的条件下进行。
在1.5mL容量的管中以以下的比例混合试剂,在25℃使其反应40小时。反应后,加入1mL丙酮并搅拌,将所产生的沈淀以12000rpm离心5分钟除去。馏去上清的丙酮,注入LC/MS进行分析。
上述粗酶提取液:300μL
化合物1的DMSO溶液:5μL
葡萄糖6磷酸溶液:5μL
NADP+溶液:5μL
葡萄糖6磷酸脱氢酶溶液:5μL
磷酸钾缓冲液(pH7.4,含有1mM EDTA):180μL。
<分析条件>
柱:カプセルパックC18MG
移动相组成:
0~3分钟:85%水、5%乙腈、10%甲酸水溶液(0.1v/v%)
3~30分钟:85→25%水、5→65%乙腈、10%甲酸水溶液(0.1v/v%)
30.1~36分钟:90%乙腈、10%甲酸水溶液(0.1v/v%)
柱温:40℃流速:0.35mL/分钟注入量:100μL
UV波长:化合物1:325nm;吡虫啉:300nm。
其结果是,代谢物的面积%的合计,化合物1为0.08,吡虫啉为2.55、与吡虫啉相比,化合物1的代谢物的量非常少。由以上的结果显示,即使对将吡虫啉代谢失活的抵抗性害虫,化合物1也可以有效地防除。
<对于动物寄生性害虫的防除效果>
参考例 长角血蜱防除试验
将本发明的化合物的200ppm、10ppm的丙酮溶液30μL加入4mL容量玻璃瓶中。将其载放于摇床上,在旋转的同时进行风干,而在瓶内壁形成化合物的干膜。将瓶干燥24小时以上,然后,在其中放饲10只长角血蜱幼螨,加盖。将瓶静置于25℃、湿度85%、全暗条件的恒温室中。放饲1日后观察其生死,依据下式算出死虫率。试验通过2连制进行。
死虫率(%)={死亡虫数/(生存虫数+死亡虫数)}×100。
其结果是,在200ppm的处理量,化合物1、化合物9显示死虫率80%以上的杀螨效果。
在10ppm的处理量,化合物1、化合物9显示死虫率80%以上的高的杀螨效果。
在同样的试验中,吡虫啉在10ppm的处理量,死虫率为4%。
参考例 小鼠体表的长角血蜱防除效果
将小鼠(ICR,雄性、5周龄)的背面体毛剃直径约2cm,在该处将切至高度约1.5cm的15mL聚苯乙烯圆锥形管用瞬间粘接剂粘接。
将用以下的处方调制的有害生物防除剂的1000倍稀释液20μL,滴加于粘接的试管内的小鼠体表。充分使其干燥后,放饲10只以上长角血蜱幼螨在管内并加盖。放饲3日后观察长角血蜱的生死,依据下式算出吸血抑制率。
处方[液化滴剂]
化合物1 48重量%
乙醇 52重量%
将上述成分均匀地混合得到液化滴剂。
吸血抑制率(%)=100-{吸血螨数/(生存螨数+死亡螨数)}×100。
其结果是,化合物1显示吸血抑制率为91%这样的长角血蜱防除效果。
产业可利用性
如以上所说明,根据本发明,可以根据需要一锅有效且收率良好地制造作为有害生物防除剂有用的所述式(I)所表示的2-酰基亚氨基吡啶衍生物,进而,可以稳定且廉价地供给作为有害生物防除剂所要求的量。因此,本发明可以较大贡献于有害生物防除的领域。
- 还没有人留言评论。精彩留言会获得点赞!