以lacZα短肽编码基因为第二基因编码框的双顺反子特异DNA及其应用的制作方法
本发明属于生物
技术领域:
,具体涉及一种以lacZα短肽编码基因为第二基因编码框的双顺反子特异DNA及其在表达目的基因中的应用。
背景技术:
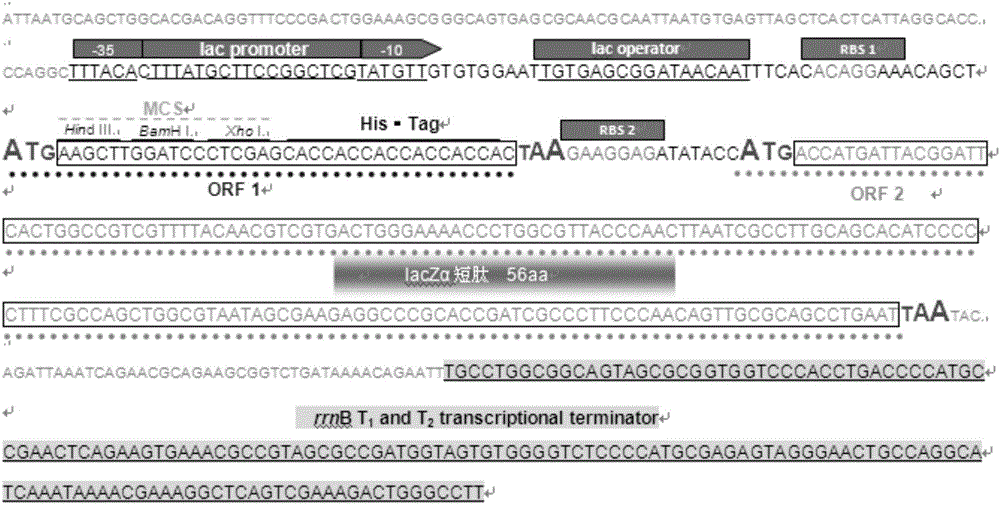
:大肠杆菌表达系统是基因表达技术中发展最早和目前应用最广的外源基因表达系统,该系统遗传背景简单,并具有使用安全、操作简便、周期短、抗污染能力强、成本低廉等其它表达系统不可比拟的优点,因而大肠杆菌被形象地称为“重组蛋白工厂”。表达载体是大肠杆菌表达系统的核心,一般一个完整的表达载体包含多个必需元件,可分为两大类。第一大类为载体元件,决定了载体质粒在宿主细胞内稳定存在的能力。包括:复制起始位点(其决定了表达载体在宿主内的拷贝数)和选择性标记(可用来筛选重组子,常用抗生素抗性标记,如氨苄青霉素抗性标记、卡那霉素抗性标记)。另一大类元件则和插入外源基因的转录及翻译有关,包括:启动子和操纵子(理想的启动子应具有强启动性,可受调节基因严谨调控)、强的转录终止序列(可高效终止转录)、优化的核糖体结合位点RBS、优化的RBS与起始密码子ATG之间的距离、可供基因工程操作的多克隆位点MCS、MCS后应有终止mRNA翻译的终止密码子。商品化的表达载体大多数都是单顺反子形式。按重组蛋白的表达方式,表达载体大致可分为:单独表达、融合表达(与伴侣蛋白、纯化标签等相融合)、分泌表达等。主要有两种公认的手段用于检测外源目的基因的表达与否及外源蛋白的水平,即蛋白质电泳和免疫印迹法。将诱导表达后的菌体进行SDS-聚丙烯酰胺凝胶电泳(SDS-PAGE)分析,相对于未诱导菌体,在理论分子量附近出现浓重的条带,可初步判断外源蛋白表达,而条带的灰度反映了蛋白表达量。进一步通过蛋白质印迹法(WesternBlot,WB)对其进一步检测,以明确其表达。可选针对外源蛋白本身的抗体作为一抗,当然大多数情况,外源蛋白均与相应表达标签融合表达,一方面利于后续纯化,另一方面可选商品化的标签抗体作为一抗,方便WB检测外源蛋白的表达。外源蛋白表达失败或不表达的原因很多,外源基因本身特性、重组蛋白细胞毒性等原因,只能通过上游的合理设计来解决。下游的操作不当或失误同样可导致外源蛋白不表达,如重组菌甘油管保存时间过长,导致质粒丢失,诱导剂加错或用量不当等,原来表达很好的重组菌突然“不表达”了。SDS-PAGE或WB检测除涉及时效性外,也经常会出现操作不当、试剂失效等原因导致的检测失败。而实验失误导致的蛋白不表达,常常需要多次重复实验进行逐一排查,费时费力。目前,尚缺乏一种方便快捷的指示外源蛋白表达的方法。技术实现要素:本发明的目的是提供一种以lacZα短肽编码基因为第二基因编码框的双顺反子特异DNA及其在表达目的基因中的应用。本发明提供了一种特异DNA分子,具有双顺反子结构,自上游至下游依次包括如下元件:lac启动区、lac操纵区、第一个核糖体结合位点、基因编码框Ⅰ、第二个核糖体结合位点、基因编码框Ⅱ、终止子;基因编码框Ⅰ的最后一个核苷酸与第二个核糖体结合位点的第一个核苷酸连接;基因编码框Ⅱ编码lacZα短肽;所述lacZα短肽由β-半乳糖苷酶(lacZ)自N末端起第1至n个氨基酸残基组成;n为50-150之间的自然数。n具体可为56。第一个核糖体结合位点与第二个核糖体结合位点,既可以为相同序列也可以为不同序列。所述lacZα短肽具体为如下(a1)或(a2):(a1)由序列表中序列5所示的氨基酸序列组成的蛋白质;(a2)将序列5的氨基酸序列经过一个或几个氨基酸残基的取代和/或缺失和/或添加且具有相同功能的由其衍生的蛋白质。所述基因编码框Ⅱ具体为如下(b1)或(b2)或(b3):(b1)序列表的序列1中自5’末端第242-412位核苷酸所示的DNA分子;(b2)在严格条件下与(b1)限定的DNA序列杂交且编码相同多肽的DNA分子;(b3)与(b1)限定的DNA序列至少具有70%、至少具有75%、至少具有80%、至少具有85%、至少具有90%、至少具有95%、至少具有96%、至少具有97%、至少具有98%或至少具有99%以上同源性且编码相同多肽的DNA分子。第一个核糖体结合位点具体如序列表的序列1中自5’末端第173-177位核苷酸所示。第二个核糖体结合位点具体如序列表的序列1中自5’末端第228-234位核苷酸所示。所述lac启动区如序列表的序列1中自5’末端第112-141位核苷酸所示。所述lac操纵区如序列表的序列1中自5’末端第146-168位核苷酸所示。所述终止子具体可为rrnBT1T2终止子。所述rrnBT1T2终止子具体可如序列表的序列1中自5’末端第458-615位核苷酸所示。基因编码框Ⅰ中可具有MCS区。MCS即多克隆位点。MCS区的作用为:方便外源目的基因的插入。基因编码框Ⅰ中还可具有多肽标签的编码序列。多肽标签的作用为:方便对外源目的基因表达的蛋白进行纯化。所述多肽标签具体可为His6标签。所述特异DNA分子具体可为如下(c1)、(c2)或(c3):(c1)序列表的序列1中自5’末端第112-615位核苷酸所示的DNA分子;(c2)序列表的序列1中自5’末端第9-621位核苷酸所示的DNA分子;(c3)序列表的序列1所示的DNA分子。含有以上任一所述特异DNA分子的重组质粒也属于本发明的保护范围。所述重组质粒中,还具有复制起始位点(ori)和抗生素抗性基因。所述抗生素抗性基因具体可为氨苄青霉素抗性基因。所述重组质粒具体可为在载体pBR322的多克隆位点(例如NdeI和EcoRI酶切位点之间)插入以上任一所述特异DNA分子得到的重组质粒。本发明还保护用于表达目的基因的试剂盒,包括以上任一所述特异DNA分子或以上任一所述重组质粒。所述试剂盒还可包括宿主菌。所述宿主菌为β-半乳糖苷酶缺陷型菌株(即lacZΔM15基因型菌株),具体可为大肠杆菌Top10、大肠杆菌DH5α或大肠杆JM109菌。所述试剂盒还可包括IPTG和/或X-gal。本发明还保护以上任一所述特异DNA分子或以上任一所述重组质粒或以上任一所述试剂盒在表达目的基因中的应用。应用所述特异DNA分子表达目的基因的方法包括如下步骤:(1)将目的基因以不改变读码框的方式插入以上任一所述特异DNA分子的基因编码框Ⅰ中(对于含有MSC区的特异DNA分子来说,插入MSC区);(2)将步骤(1)得到的DNA分子导入以上任一所述宿主菌,得到重组菌;(3)培养所述重组菌并在培养过程中加入IPTG和X-gal,通过对培养体系或培养上清进行观察或检测判断目的基因是否表达。应用所述重组质粒表达目的基因的方法包括如下步骤:(1)将目的基因以不改变读码框的方式插入以上任一所述重组质粒中的特异DNA分子的基因编码框Ⅰ中(对于含有MSC区的特异DNA分子来说,插入MSC区);(2)将步骤(1)得到的质粒导入以上任一所述宿主菌,得到重组菌;(3)培养所述重组菌并在培养过程中加入IPTG和X-gal,通过对培养体系或培养上清进行观察或检测判断目的基因是否表达。通过对培养体系进行观察判断目的基因是否表达的方法如下:观察培养体系的颜色,如果培养体系显示为蓝色说明目的基因表达(培养体系的蓝色越深说明目的基因的表达越多),如果培养体系不显示为蓝色说明目的基因未表达。通过对培养体系进行检测判断目的基因是否表达的方法如下:检测培养上清的OD630nm值,如果OD630nm值为0以上说明目的基因表达(OD630nm值越大说明目的基因表达越多),如果OD630nm值为0说明目的基因未表达。通常认为核糖体遇终止密码子将发生大小亚基解离,本发明提供的特异DNA分子中,基因编码框Ⅰ中的终止密码子后紧跟着第二个核糖体结合位点,则部分核糖体将来不及解离而接下去启动了基因编码框Ⅱ的翻译,即报告系统被激活,从而实现了指示外源基因表达的能力。如果核糖体在此之前遇到了表达阻力,而使得基因编码框Ⅰ不表达,或表达意外终止,将不会启动基因编码框Ⅱ的翻译,即报告系统无法激活,提示表示失败。完整的大肠杆菌β-半乳糖苷酶基因编码由1024个氨基酸残基组成的蛋白质,如果利用完整的β-半乳糖苷酶构建报告系统,大肠杆菌细胞将消耗过多的能量在β-半乳糖苷酶的表达上,不利于目的基因的高表达。此外,完整的β-半乳糖苷酶编码序列高达3096bp,如此大的片断在表达载体的构建上也存在一定的技术难度。本发明提供的特异DNA分子中,以lacZα短肽的编码基因为基因编码框Ⅱ,极大地节约了大肠杆菌表达所需的能量及原料,对基因编码框Ⅰ的表达的影响微乎其微。宿主菌为β-半乳糖苷酶缺陷型菌株,其基因组中编码β-半乳糖苷酶突变体,缺失了N端的部分氨基酸残基,无酶活性。β-半乳糖苷酶突变体与lacZα短肽互补,形成完整的有活性的β-半乳糖苷酶,可水解底物X-gal生成蓝色化合物,便于裸眼直接观察。应用所述特异DNA分子表达目的基因时,将目的基因以不改变读码框的方式插入基因编码框Ⅰ中,然后导入宿主菌。在IPTG诱导下,转录生成一条双顺反子融合mRNA。只有在外源目的基因成功翻译后,才能启动lacZα短肽编码序列的翻译。因此,可以通过β-半乳糖苷酶活性的检测实现目的基因的表达情况的实时监测。本发明提供的DNA分子中,采用lacZα短肽指示外源目的基因的表达情况。在诱导表达的同时,不借助任何仪器设备就能判断外源目的基因的表达与否,不需要额外的SDS-PAGE或WB检测。本发明还保护一种特异DNA分子,具有双顺反子结构,自上游至下游依次包括如下元件:第一个核糖体结合位点、基因编码框Ⅰ、第二个核糖体结合位点、基因编码框Ⅱ;基因编码框Ⅰ的最后一个核苷酸与第二个核糖体结合位点的第一个核苷酸连接。本发明提供的特异DNA分子,无需特殊步骤直接观察即可判断基因编码框Ⅰ内的外源目的基因表达与否、外源目的基因表达量的双重能力,具有重大的应用价值。附图说明图1为载体pBR322的结构示意图。图2为序列表的序列1中,NdeI识别序列与EcoRI识别序列之间的核苷酸序列的元件示意图。图3为各个重组质粒的元件示意图。图4为实施例3的结果。具体实施方式以下的实施例便于更好地理解本发明,但并不限定本发明。下述实施例中的实验方法,如无特殊说明,均为常规方法。下述实施例中所用的试验材料,如无特殊说明,均为自常规生化试剂商店购买得到的。以下实施例中的定量试验,均设置三次重复实验,结果取平均值。基因编码框:基因中起始密码子至终止密码子之间的区域(含起始密码子和终止密码子,可含有内含子也可不含有内含子)。RBS即核糖体结合位点。大肠杆菌Top10:Invitrogen公司,产品目录号为C404010。载体pBR322(结构示意图见图1):生工生物工程(上海)有限公司,产品目录号为B610010。实施例1、重组质粒的构建1、合成序列表的序列1所示的双链DNA分子。序列表的序列1中,第9-14位核苷酸为限制性内切酶NdeI的识别序列,第112-141位核苷酸为lac启动区,第146-168位核苷酸为lac操纵区,第173-177位核苷酸为RBS1,第186-188位核苷酸为基因编码框Ⅰ的起始密码子,第189-206位核苷酸为MCS区(依次为HindⅢ酶切识别序列、BamHⅠ酶切识别序列和XhoⅠ酶切识别序列),第207-224位核苷酸为His6标签的编码序列,第225-227位核苷酸为基因编码框Ⅰ的终止密码子,第228-234位核苷酸为RBS2,第242-412位核苷酸为lacZα短肽的编码序列(第242-244位核苷酸为基因编码框Ⅱ的起始密码子,第410-412位核苷酸为基因编码框Ⅱ的终止密码子),第458-615位核苷酸为rrnBT1T2终止子,第616-621位核苷酸为限制性内切酶EcoRI的识别序列。序列表的序列1中,NdeI识别序列与EcoRI识别序列之间的核苷酸序列的元件示意图见图2。2、取步骤1得到的DNA分子,用限制性内切酶NdeI和EcoRI双酶切,回收酶切产物。3、取载体pBR322,用限制性内切酶NdeI和EcoRI双酶切,回收约2063bp的DNA片段(包含ori区和氨苄青霉素抗性基因)。4、将步骤2得到的酶切产物与步骤3得到的DNA片段连接,得到重组质粒pBR-ORF-lacZα。经测序,重组质粒pBR-ORF-lacZα中具有序列表的序列1第9-621位核苷酸所示的DNA分子,也具有ori区和氨苄青霉素抗性基因。5、将序列表的序列2第7-610位核苷酸所示的双链DNA分子插入载体pBR322的NdeI和EcoRI酶切位点之间,得到重组质粒pBR-ORFmu1-lacZα。经测序,重组质粒pBR-ORFmu1-lacZα与重组质粒pBR-ORF-lacZα的差异仅在于在用序列表的序列2所示DNA分子取代了序列表的序列1第9-621位核苷酸所示的DNA分子。序列2与序列1第9-621位的差别在于插入了1个稀有密码子“CTA”。6、将序列表的序列3第7-613位核苷酸所示的双链DNA分子插入载体pBR322的NdeI和EcoRI酶切位点之间,得到重组质粒pBR-ORFmu2-lacZα。经测序,重组质粒pBR-ORFmu2-lacZα与重组质粒pBR-ORF-lacZα的差异仅在于在用序列表的序列3所示DNA分子取代了序列表的序列1第9-621位核苷酸所示的DNA分子。序列3与序列1第9-621位的差别在于插入了2个稀有密码子“CTAATA”。7、将序列表的序列4第7-616位核苷酸所示的双链DNA分子插入载体pBR322的NdeI和EcoRI酶切位点之间,得到重组质粒pBR-ORFmu3-lacZα。经测序,重组质粒pBR-ORFmu3-lacZα与重组质粒pBR-ORF-lacZα的差异仅在于在用序列表的序列4所示DNA分子取代了序列表的序列1第9-621位核苷酸所示的DNA分子。序列4与序列1第9-621位的差别在于插入了3个稀有密码子“CTAATAAGG”。实施例2、构建重组菌将重组质粒pBR-ORF-lacZα导入大肠杆菌Top10,得到重组菌Ⅰ。将重组质粒pBR-ORFmu1-lacZα导入大肠杆菌Top10,得到重组菌Ⅱ。将重组质粒pBR-ORFmu2-lacZα导入大肠杆菌Top10,得到重组菌Ⅲ。将重组质粒pBR-ORFmu3-lacZα导入大肠杆菌Top10,得到重组菌Ⅳ。各个重组质粒的元件示意图见图3。稀有密码子,特别是连续的稀有密码子,对于基因表达存在阻力。当第一个基因编码框内出现稀有密码子时,相比无稀有密码子的情况,第二个基因编码框的翻译水平将降低,即第二个基因编码框内的报告基因的表达水平将明显降低,体现为β-半乳糖苷酶活性降低,进一步表现出为水解X-gal生成蓝色产物的能力下降。当第一个基因编码框内出现3个连续的稀有密码子时,其翻译将提前终止,核糖体大小亚基提前解离,导致第二个基因编码框将无法启动翻译,体现为无β-半乳糖苷酶产生,进而无法水解X-gal,菌液不变色。实施例3、通过颜色或OD值检测重组菌中基因编码框Ⅰ的表达情况一、试验处理分别将实施例2构建的重组菌Ⅰ、重组菌Ⅱ、重组菌Ⅲ和重组菌Ⅳ作为待测菌,进行如下操作:1、将待测菌接种至含50μg/mL氨苄青霉素的液体LB培养基中,37℃、250rpm振荡培养至OD600nm值=0.6。2、完成步骤1后,在培养体系中加入IPTG和X-gal(IPTG在培养体系中的浓度为0.5mM,X-gal在培养体系中的浓度为0.04g/100ml),37℃、250rpm振荡培养4小时。3、完成步骤2后,取整个培养体系,3500rpm离心,取上清液,拍照。4、取步骤3得到的上清液,检测630nm吸光度。二、对照处理1、将重组菌Ⅰ接种至含50μg/mL氨苄青霉素的液体LB培养基中,37℃、250rpm振荡培养至OD600nm值=0.6。2、完成步骤1后,继续37℃、250rpm振荡培养4小时。3、完成步骤2后,取整个培养体系,3500rpm离心,取上清液,拍照。4、取步骤3得到的上清液,检测630nm吸光度。三、结果分析上清液的照片见图4。图4中,1为试验处理中重组菌Ⅰ得到的上清液,2为试验处理中重组菌Ⅱ得到的上清液,3为试验处理中重组菌Ⅲ得到的上清液,4为试验处理中重组菌Ⅳ得到的上清液,5为对照处理中重组菌Ⅰ得到的上清液。可以观察到,图4中1、2、3的颜色为蓝色,但颜色依次变浅,4和5不显示蓝色。结果表明,随着引入第一个基因编码框内的稀有密码子的数量上升,上清液蓝色程度逐渐下降;引入连续3个稀有密码子后,上清液肉眼已观察不到蓝色出现,与未诱导菌液上清颜色基本一致。上清液的OD630nm值见表1。与图4的结果一致。表1OD630nm值试验处理中重组菌Ⅰ得到的上清液0.121试验处理中重组菌Ⅱ得到的上清液0.085试验处理中重组菌Ⅲ得到的上清液0.032试验处理中重组菌Ⅳ得到的上清液0对照处理中重组菌Ⅰ得到的上清液0当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 蝎神经毒素AaIT及其编码基因与应用的制造方法与工艺
- 腈水解酶突变体及其编码基因和应用的制造方法与工艺
- 植物抗旱相关蛋白AsTMK1及其编码基因和应用的制造方法与工艺
- 植物抗旱相关蛋白EtSnRK2.2及其编码基因和应用的制造方法与工艺
- OsSAPK7蛋白及其编码基因在提高水稻白叶枯病抗性中的应用的制造方法与工艺
- 一种抗除草剂抗虫融合基因、编码蛋白及应用的制造方法与工艺
- 毛果杨PtrZFP103基因及其编码蛋白和应用的制造方法与工艺
- 大豆bHLH转录因子基因GmFER及其编码蛋白和应用的制造方法与工艺
- 一种花狭口蛙的基因及其编码的多肽和该多肽的用途的制造方法与工艺
- 一种泽陆蛙基因及其编码的多肽和该多肽的用途的制造方法与工艺