一种烟草腋芽生长调控基因NtBRC1及其克隆方法与应用与流程
本发明属于遗传工程技术领域,具体涉及一种烟草腋芽生长调控基因NtBRC1及其克隆方法与应用。
背景技术:
烟草(Nicotiana tabacum)是重要的经济作物,是茄科一年生草本植物。打顶抹芽是优质烤烟种植过程中一项重要的生产技术措施。烟草在一定的生长时期必须打顶,而打顶后会萌生大量的侧芽。侧芽耗费营养,会降低烟叶的品质和产量,因此必须抹掉侧芽。人工抹芽耗费人力和物力,提高了烟叶生产成本。打顶后施用化学药剂可抑制腋芽生长,降低了烟叶生产劳动强度。但化学药剂需要成本,且会造成环境污染。用发明以发现打顶后产生侧芽(腋芽)的相关基因为突破口,通过生物技术创建无侧芽、少侧芽或小侧芽的烟草,则有可能从根本上或大幅度解决上述问题,从而实现 烟草生长量、烟叶品质、环保等方面均有较大提升。
BRC1是一个TCP家族的转录因子蛋白基因。其在控制分枝生长中的功能已有大量报道,BRC1基因突变的拟南芥比野生型莲座增加;豌豆BRC1基因突变后分枝增加。我们对烟草NtBRC1基因也进行了广泛理论研究和应用实践,发现NtBRC1在烟草打顶后具有促进侧芽(腋芽)生长的功能。通过植物基因工程技术控制烟草打顶后侧芽生长发育方面具有生产应用重要意义。
国际上一些知名烟草公司和研究机构正采用生物技术和基因工程技术对烟草无腋芽少腋芽品种进行研究,以提高烟草品质,减少对烟草打顶后化学药剂的施用,以降低烟叶生产劳动强度、化学药剂成本和环境污染。目前,我国也提出了无腋芽优质烟的开发计划,为烟草无腋芽少腋芽品种研究提供了无限契机。随着烟草无腋芽少腋芽品种研究的深入,必将使我国烟草腋芽控制迈上新台阶,为我国烟叶的健康和可持续发展奠定基础。因此,开发一种利用生物技术手段创制无腋芽少腋芽品种的候选基因,通过基因操作减少烟草腋芽是非常必要的。
技术实现要素:
本发明的第一目的在于提供一种烟草腋芽生长调控基因NtBRC1;第二目的在于提供所述的烟草腋芽生长调控基因NtBRC1的克隆方法;第三目的在于提供所述的烟草腋芽生长调控基因NtBRC1的应用。
本发明的第一目的是这样实现的,所述的烟草腋芽生长调控基因NtBRC1的核苷酸序列如SEQ ID NO:1所示。
本发明的第二目的是这样实现的,包括以下步骤:
A、确定NtBRC1基因序列;
(1)利用番茄基因SIBRC1b搜索烟草基因组数据库得到烟草NtBRC1的核苷酸序列,利用此序列设计基因克隆引物:
正向引物:5′- ATGTATCCGCCAAGCAACAG -3′;
反向引物:5′- GCTATTGAAATCCTAAAAAAT -3′;
B、提取烟草根组织RNA,反转录得到第一链cDNA;
C、根据NtBRC1基因序列设计合成特异性引物,以反转录得到的第一链cDNA作为模板,进行PCR扩增,选用Phusion高保真扩增酶反应体系,体系总体积50μL,包括:200ng cDNA,5×Phusion HF反应缓冲液,10mM dNTP,2U的Phusion® High-Fidelity DNA Polymerase,正反向引物各1μM,PCR反应在Mastercycler® pro扩增仪上进行,反应程序为:98℃,30秒;98℃,7秒,55℃,30秒,72℃,30秒,35个循环;72℃延伸7分钟;回收和纯化PCR产物;
D、纯化产物与载体连接,通过试剂盒反应与TOPO载体连接,连接体系与过程如下: 4μL纯化产物+1μL salt salution +1μL PCR-BluntⅡ-TOPO 混匀,25℃,水浴30min;将连好的载体通过热激转化进入大肠杆菌,加液体培养基震荡培养后涂到含100mg/L 卡那霉素的LB平板上过夜培养, 长出菌后挑取菌落进行培养,培养后取2ml菌液提取质粒,进行质粒PCR检测,筛选阳性克隆,对阳性克隆进行测序。
本发明的第三目的是这样实现的,所述的烟草腋芽生长调控基因NtBRC1在获得降低打顶后烟草产生腋芽的发育长度的转基因植株中的应用。
本发明利用同源克隆技术从烟草中得到一个烟草腋芽生长调控基NtBRC1,具体步骤为:利用利用番茄基因SIBRC1b(GenBank登录号: HM597230)搜索烟草基因组数据库(https://solgenomics.net/)得到烟草NtBRC1的核苷酸序列。利用此序列设计基因克隆引物;利用此序列信息设计基因特异引物进行PCR反应,得到目的产物;对目的产物测序,得到NtBRC1基因序列;利用农杆菌介导的遗传转化方法获得NtBRC1基因的过表达(OE)植株,对NtBRC1进行功能验证,结果表明NtBRC1基因具有控制烟草侧芽生长发育的功能。NtBRC1基因的发现,为通过调控基因的表达,控制烟草侧芽生长发育,为生产绿色优质烟叶提供了基因资源。
附图说明
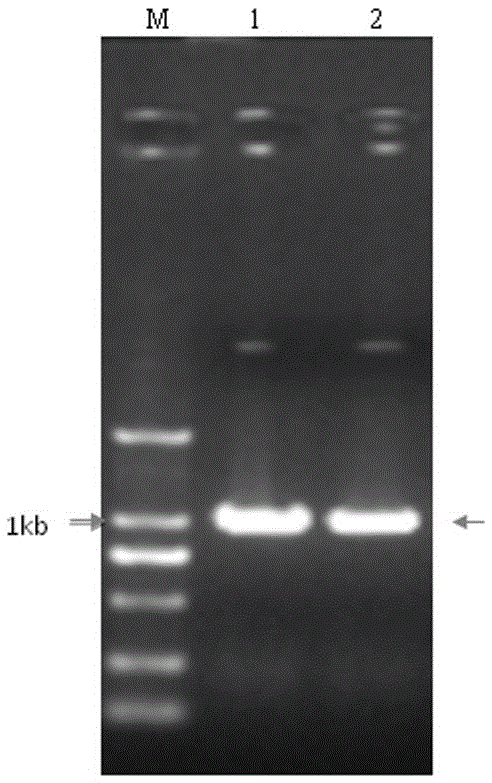
图1为利用引物对NtBRC1F/NtBRC1R扩增NtBRC1基因结果,扩增产物987bp;
图2为TOPO-NtBRC1载体PCR鉴定,所用引物对为NtBRC1F/NtBRC1R,扩增产物为987 bp;
图3为 TOPO-NtBRC1载体酶切鉴定,利用BamHI和XhoI双酶切,可以获得1000bp左右的目的条带;
图4为 NtBRC1基因过表达(OE)载体目标片段PCR扩增结果,所用引物对为NtBRC1-OE-F / NtBRC1-OE-R,扩增产物为999 bp;
图5为过表达(OE)载体的酶切验证,BamHI和XhoI双酶切后,可以将1000bp左右的目的条带切下;
图6为NtBRC1过表达(OE)烟草腋芽生长长度,Vector Control(VC)为转空载体对照,其余为转基因株系。
具体实施方式
下面结合实施例和附图对本发明作进一步的说明,但不以任何方式对本发明加以限制,基于本发明教导所作的任何变换或替换,均属于本发明的保护范围。
本发明所述的烟草腋芽生长调控基因NtBRC1的核苷酸序列如SEQ ID NO:1所示。
所述的烟草腋芽生长调控基因NtBRC1编码的氨基酸序列如SEQ ID NO:2所示。
本发明所述的烟草腋芽生长调控基因NtBRC1的克隆方法,其特征在于包括以下步骤:
A、确定NtBRC1基因序列;
(1)利用番茄基因SIBRC1b搜索烟草基因组数据库得到烟草NtBRC1的核苷酸序列,利用此序列设计基因克隆引物:
正向引物:5′- ATGTATCCGCCAAGCAACAG -3′;
反向引物:5′- GCTATTGAAATCCTAAAAAAT -3′;
B、提取烟草根组织RNA,反转录得到第一链cDNA;
C、根据NtBRC1基因序列设计合成特异性引物,以反转录得到的第一链cDNA作为模板,进行PCR扩增,选用Phusion高保真扩增酶反应体系,体系总体积50μL,包括:200ng cDNA,5×Phusion HF反应缓冲液,10mM dNTP,2U的Phusion® High-Fidelity DNA Polymerase,正反向引物各1μM,PCR反应在Mastercycler® pro扩增仪上进行,反应程序为:98℃,30秒;98℃,7秒,55℃,30秒,72℃,30秒,35个循环;72℃延伸7分钟;回收和纯化PCR产物;
D、纯化产物与载体连接,通过试剂盒反应与TOPO载体连接,连接体系与过程如下: 4μL纯化产物+1μL salt salution +1μL PCR-BluntⅡ-TOPO混匀,25℃,水浴30min;将连好的载体通过热激转化进入大肠杆菌,加液体培养基震荡培养后涂到含100mg/L 卡那霉素的LB平板上过夜培养, 长出菌后挑取菌落进行培养,培养后取2ml菌液提取质粒,进行质粒PCR检测,筛选阳性克隆,对阳性克隆进行测序。
C步骤中所述的特异性引物引物为:
正向引物:5′- ATGTATCCGCCAAGCAACAG -3′;
反向引物:5′- GCTATTGAAATCCTAAAAAAT -3′。
D步骤中所述的培养基为:称取胰蛋白胨10g,酵母提取物5g,NaCl 10g 溶解于1L 蒸馏水中,高温灭菌,即121℃,25min灭菌后使用。
本发明所述的烟草腋芽生长调控基因NtBRC1的应用为所述的烟草腋芽生长调控基因NtBRC1在获得降低打顶后烟草产生腋芽的发育长度的转基因植株中的应用。
所述获得NtBRC1转基因烟草的方法包括以下步骤:
A、构建过表达(OE)载体:
a、以pCR®-BluntII-TOPO载体为中间载体、pK2G7为表达载体构建NtBRC1基因的过表达(OE)载体,构建引物为:
NtBRC1-OE-F: 5’- GGATCCATGTATCCGCCAAGCAACAG -3’,
NtBRC1- OE-R: 5’- CTCGAGCTATTGAAATCCTAAAAAAT -3’,
NtBRC1- OE-F中GGATCC处为BamH I酶切位点;NtBRC1-OE-R中CTCGAG处为XhoI酶切位点;
b、以NtBRC1阳性克隆TOPO质粒DNA为模板进行PCR扩增,PCR反应体积为50μL,包括:200ng cDNA,5×Phusion HF反应缓冲液,10mM dNTP,2U的Phusion® High-Fidelity DNA Polymerase,NtBRC1- OE-F引物、NtBRC1- OE-R引物各1μM,PCR反应在Mastercycler® pro扩增仪上进行,反应程序为:98℃,30秒;98℃,7秒,55℃,30秒,72℃,30秒,30个循环;72℃延伸7分钟;回收和纯化PCR产物;
c、回收目的片段产物与TOPO载体连接:通过试剂盒反应与TOPO载体连接。连接体系与过程如下: 4μL纯化产物+1μL salt salution +1μL PCR®-BluntⅡ-TOPO混匀,25℃,水浴30min,将连好的载体通过热激转化进入大肠杆菌,加培养基震荡培养后涂到含100mg/L 卡那霉素的LB平板上过夜培养, 长出菌后挑取菌落进行培养,培养后取2ml菌液提取质粒,进行质粒PCR检测,筛选阳性克隆,对阳性克隆进行测序;
d、入门克隆pENTRTM2B-NtBRC1OE的构建:用上步测序对的TOPO质粒和pENTRTM2B空载体做酶切反应:BamH I+XhoI双酶切TOPO质粒和pENTRTM2B-ccdB得到目的基因片段和载体片段pENTRTM2B,切胶回收后进行连接、连接体系:连接反应总体积为10μL,含酶切目的片段的切胶回收产物4μL,pENTRTM2B(BamHI+XhoI双酶切)载体1μL,10×T4 buffer 1μL,T4 Ligase 1μL,灭菌双蒸水3μL,混匀,室温反应2小时,转化入大肠杆菌感受态细胞,加培养基震荡培养后涂到含100mg/L 卡那霉素的LB平板上过夜培养,提取质粒进行PCR检测和酶切检测,以验证目的基因片段是否已经插入载体片段,入门载体是否构建成功;
e、通过LR反应得到植物表达载体:挑选检测正确的入门克隆与植物表达载体PK2G7 (Destination clone)进行LR反应,具体反应如下:体系与过程如下:
1)连上2B载体的入门克隆(50-150ng )1-7μL + 0.5μL Destination Vector + TE Buffer(up to 8μL);
2)上述体系混匀,冰浴2min,轻弹2次;
3)加入2μL LR CloneaseTMⅡ enzyme Mix 到上述体系,轻弹,混匀,离心,25℃水浴1h;
4)加入1μL Proteinase K 轻弹,混匀,37℃水浴10min;
通过热激转化转入大肠杆菌,加入培养基震荡培养后涂含100mg/L 壮观霉素的LB平板上,得到植物表达载体,挑菌提质粒后,进行PCR验证及酶切验证。PCR反应体积为25μL,包括:100ng质粒DNA,5×Phusion HF反应缓冲液,10mM dNTP,1U的Phusion® High-Fidelity DNA Polymerase,NtBRC1-OE-F引物、NtBRC1-OE-R引物各0.5μM,PCR反应在Mastercycler® pro扩增仪上进行,反应程序为:98℃,30秒;98℃,7秒,55℃,30秒,72℃,30秒,30个循环;72℃延伸7分钟;检测重组质粒的正确性,理论上基因上下游引物扩增的片段大小为987bp,验证PCR扩增结果是否和预期一致;酶切体系为20μL:包含质粒DNA1μg,BamHI、XhoI各0.5μL,NEB Buffer 3.1 2μL,理论上酶切检测结果分别为载体片段约12kb和目的基因片段999bp,同时满足两个条件,则表明表达载体构建成功,将检测对的质粒进行测序比对,进一步验证;
B、农杆菌转化:
从-80℃冰箱中取出农杆菌感受态细胞,在冰上溶解,并加入重组表达载体PK7G- NtBRC1OE 4μL;将混合物分别放入液氮速冻1分钟,转入37℃水浴5分钟,再冰浴2分钟,向混合物中加入1mL LB液体培养基,28℃、220rpm培养3~4小时;培养物涂布于含有壮观霉素100mg/L和利福平25mg/L的LB固体培养基上,28℃倒置培养2~3天,可见含有目的载体的农杆菌克隆;
C、过表达(OE)载体导入烟草、培养转基因植株:
a、挑取含有目标载体的农杆菌克隆,在含有壮观霉素和利福平的LB平板上划线,28℃培养2~3天;刮取划线菌斑接菌于含有壮观霉素和利福平的MS培养基中,28℃,220rpm震荡培养,菌液浓度达到OD=0.5~0.8时进行侵染,得到含目标载体的农杆菌菌体;将菌体收集到离心管中,6,000rpm离心5分钟富集菌体,弃上清,再用20ml液体MS培养基重悬菌体,得到含目标载体的农杆菌悬浮菌液;
b、将烟草叶片置于500mL广口瓶中, 加入适量75%乙醇,漂洗1min;倒掉乙醇,加入0.1%的HgCl2溶液,置摇床上室温振荡15~30分钟;倒掉溶液,用无菌水冲洗6遍;
c、将烟草叶片取出,用灭菌吸水纸洗去表面液体,取无菌叶片用剪刀切成1cm×1cm的小片,将切成小片的烟草叶片分别放入含目标载体的无菌MS液体培养基悬浮菌液中,静置15~20min;取出烟草叶片,用无菌滤纸吸去多余菌液,于加入6-BA 1.0mg/L、NAA 0.1mg/L的MS培养基中25℃暗培养两天;把烟草叶片转入分化培养基中,切口接触培养基,于温室条件下分化培养;分化培养基为加入6-BA 1.0mg/L、NAA 0.1mg/L、Kan 100mg/L、头孢霉素500mg/L的MS培养基,每2~3周将其转移到新的培养基中,切口处逐渐长出愈伤组织,最后分化出芽;
d、将长至3~5cm的芽切下,转入MS培养基诱导生根,生根后的转基因植株由生根培养基中取出,用自来水净培养基,移植于灭菌的营养土中;
e、转基因植株经NPTII基因特异引物PCR验证扩增,鉴定转基因阳性植株。
C步骤b中所述的0.1%的HgCl2溶液配制方法为称取升汞0.1克,用少许酒精溶解,再加水定容至100毫升。
下面以具体实施案例对本发明做进一步说明:
实施例1 ——NtBRC1基因的分离克隆
利用番茄基因SIBRC1b(GenBank登录号: HM597230)搜索烟草基因组数据库(https://solgenomics.net/)得到烟草NtBRC1的核苷酸序列。根据该序列设计克隆基因特异引物进行PCR反应,得到目的产物;对目的产物测序,得到NtBRC1基因序列;其核苷酸序列为SEQ ID NO.1所示,编码328个氨基酸,如序列表SEQIDNO.2所示。
提取云烟87根部RNA,抽提使用Trizol试剂盒(Invitrogen,按该试剂盒提供的说明书操作)。
取2μg RNA进行反转录,利用PrimeScript™ RT reagent Kit with gDNA Eraser(TAKARA)试剂盒进行基因组DNA去除并反转录(按试剂盒操作手册操作)。
将反转录得到的第一链cDNA作为模板,用于PCR扩增NtBRC1基因全长,并设计特性行引物如下:
正向引物:NtBRC1F:5′- ATGTATCCGCCAAGCAACAG -3′;
反向引物:NtBRC1R:5′- GCTATTGAAATCCTAAAAAAT -3′。
PCR反应体系为50μL,包括:200ng cDNA,5×Phusion HF反应缓冲液,10mM dNTP,2U的Phusion® High-Fidelity DNA Polymerase,上述引物各1μM;PCR反应在Mastercycler® pro(Eppendorf,德国)扩增仪上进行,反应程序为:98℃,30秒;98℃,7秒,55℃,30秒,72℃,30秒,35个循环;72℃延伸7分钟。
产物经1%(g/mL)的琼脂糖凝胶电泳分离,扩增结果产生一条单一PCR条带,见图1。用QIAquick Gel Extraction Kit(QIAGEN,德国)回收扩增条带,提取步骤参照试剂盒使用说明。回收纯化的DNA与TOPO载体(Invitrogen ZERO BluntII -TOPO -PCR Cloning kit)连接,按说明书操作。连接产物利用热激法转化大肠杆菌,在含有100mg/L卡那霉素的LB固体平板中筛选阳性克隆,挑取5个克隆测序(大连宝生物工程有限公司)。
测序结果表明NtBRC1基因的全长为987bp,其核苷酸序列为SEQ ID NO:1所示,通过测序、比对确定是本发明需要的目的基因,申请人将该基因命名为NtBRC1。NtBRC1基因包括987bp的开放阅读框;编码328个氨基酸。
实施例2 ——过表达(OE)载体构建
a、以PCR®-BluntII-TOPO载体为中间载体、PK2G7为表达载体构建NtBRC1基因的过表达(OE)载体,构建引物为:
NtBRC1-OE-F: 5’- GGATCCATGTATCCGCCAAGCAACAG -3’,
NtBRC1-OE-R: 5’- CTCGAGCTATTGAAATCCTAAAAAAT -3’,
NtBRC1-OE-F中GGATCC处为BamH I酶切位点;NtBRC1-OE-R中CTCGAG处为Xho I酶切位点;
b、以NtBRC1阳性克隆TOPO质粒DNA为模板进行PCR扩增,PCR反应体积为50μL,包括:200ng cDNA,5×Phusion HF反应缓冲液,10mM dNTP,2U的Phusion® High-Fidelity DNA Polymerase, NtBRC1-OE-F引物、NtBRC1-OE-R引物各1μM,PCR反应在Mastercycler® pro扩增仪上进行,反应程序为:98℃,30秒;98℃,7秒,55℃,30秒,72℃,30秒,30个循环;72℃延伸7分钟;回收和纯化PCR产物;
c、回收目的片段产物与TOPO载体连接:通过试剂盒反应与Topo载体连接。连接体系与过程如下:4μL纯化产物+1μL salt salution +1μL PCR®-BluntⅡ-TOPO混匀,25℃,水浴30min。将连好的载体通过热转化进入大肠杆菌,加培养基震荡培养后涂到含100mg/L 卡那霉素的LB平板上过夜培养。 长出菌后挑取菌落进行培养,培养后取2ml菌液提取质粒,进行质粒PCR检测。筛选阳性克隆,对阳性克隆进行测序;
d、入门克隆pENTRTM2B-NtBRC1 OE的构建:用上步测序对的TOPO质粒和pENTRTM2B空载体做酶切反应:BamH I+XhoI双酶切TOPO质粒和pENTRTM2B-ccdB得到目的基因片段和载体片段pENTRTM2B,切胶回收后进行连接、连接体系:连接反应总体积为10μL,含酶切目的片段的切胶回收产物4μL,pENTRTM2B(BamH I+XhoI双酶切)载体1μL,10×T4 buffer 1μL,T4 Ligase 1μL,灭菌双蒸水3μL,混匀,室温反应2小时,转化入大肠杆菌感受态细胞,加培养基震荡培养后涂到含100mg/L 卡那霉素的LB平板上过夜培养,提取质粒进行PCR检测和酶切检测,以验证目的基因片段是否已经插入载体片段,入门载体是否构建成功。
e、通过LR反应得到植物表达载体:挑选检测正确的入门克隆与植物表达载体PK2G7 (Destination clone)进行LR反应,具体反应如下:体系与过程如下:
1)连上2B载体的入门克隆(50-150ng )1-7μL + 0.5μL Destination Vector + TE Buffer(up to 8μL)
2)上述体系混匀,冰浴2min,轻弹2次
3)加入2μL LR CloneaseTMⅡ enzyme Mix 到上述体系,轻弹,混匀,离心,25℃水浴1h。
4)加入1μL Proteinase K 轻弹,混匀,37℃水浴10min 。
通过热激转化转入大肠杆菌,加入培养基震荡培养后涂含100mg/L 壮观霉素的LB平板上。得到植物表达载体,挑菌提质粒后,进行PCR验证及酶切验证。PCR反应体积为25μL,包括:100ng质粒DNA,5×Phusion HF反应缓冲液,10mM dNTP,1U的Phusion® High-Fidelity DNA Polymerase, NtBRC1-OE-F引物、NtBRC1-OE-R引物各0.5μM,PCR反应在Mastercycler® pro扩增仪上进行,反应程序为:98℃,30秒;98℃,7秒,55℃,30秒,72℃,30秒,30个循环;72℃延伸7分钟;检测重组质粒的正确性,理论上基因上下游引物扩增的片段大小为987bp,验证PCR扩增结果是否和预期一致;酶切体系为20μL:包含质粒DNA1μg,BamH I、XhoI各0.5μL,NEB Buffer 3.1 2μL,理论上酶切检测结果分别为载体片段约12kb和目的基因片段999bp。同时满足两个条件,则表明表达载体构建成功,将检测对的质粒进行测序比对,进一步验证。
实施例3——烟草的遗传转化
(1)表达载体转化农杆菌
从-80℃冰箱中取出农杆菌感受态细胞,在冰上溶解,并加入重组表达载体PK2G7- NtBRC1 4μL;将混合物分别放入液氮速冻1分钟,转入37℃水浴5分钟,再冰浴2分钟,向混合物中加入1mL LB液体培养基,28℃、220rpm培养3~4小时;培养物涂布于含有壮观霉素100mg/L和利福平25mg/L的LB固体培养基上,28℃倒置培养2~3天,可见含有目的载体的农杆菌克隆;
(2)烟草转化
a、挑取含有目标载体的农杆菌克隆,在含有壮观霉素和利福平的LB平板上划线,28℃培养2~3天;刮取划线菌斑接菌于含有壮观霉素和利福平的MS培养基中,28℃,220rpm震荡培养,菌液浓度达到OD=0.5~0.8时进行侵染,得到含目标载体的农杆菌菌体;将菌体收集到离心管中,6,000rpm离5心分钟富集菌体,弃上清,再用20ml液体MS培养基重悬菌体,得到含目标载体的农杆菌悬浮菌液;
b、将烟草叶片置于500mL广口瓶中, 加入适量75%乙醇,漂洗1min;倒掉乙醇,加入0.1%的HgCl2溶液,置摇床上室温振荡15~30分钟;倒掉溶液,用无菌水冲洗6遍;
c、将烟草叶片取出,用灭菌吸水纸洗去表面液体,取无菌叶片用剪刀切成1cm×1cm的小片,将切成小片的烟草叶片分别放入含目标载体的无菌MS液体培养基悬浮菌液中,静置15~20min;取出烟草叶片,用无菌滤纸吸去多余菌液,于加入6-BA 1.0mg/L、NAA 0.1mg/L的MS培养基中25℃暗培养两天;把烟草叶片转入分化培养基中,切口接触培养基,于温室条件下分化培养;分化培养基为加入6-BA 1.0mg/L、NAA 0.1mg/L、Kan 100mg/L、头孢霉素500mg/L的MS培养基,每2~3周将其转移到新的培养基中,切口处逐渐长出愈伤组织,最后分化出芽;
d、将长至3~5cm的芽切下,转入MS培养基诱导生根,生根后的转基因植株由生根培养基中取出,用自来水净培养基,移植于灭菌的营养土中;
e、转基因植株经NPTII基因特异引物PCR验证扩增,鉴定转基因阳性植株。
实施例4 ——转基因植株打顶试验
转基因植株移栽到一次性花盆后,生长16天后,打顶,测定各腋芽的长度,结果见图6。过表达(OE)植株比空载体对照Vector Control(VC)降低约60-75%。上述结果表明,NtBRC1基因负责控制烟草腋芽生长的长度通过对该基因的遗传操作,可以降低烟草腋芽的生长,该基因的开发利用有利于减少烟叶生产中抑芽剂的使用。
SEQUENCE LISTING
<110> 云南省烟草农业科学研究院
<120> 一种烟草腋芽生长调控基因NtBRC1及其克隆方法与应用
<130> 2016
<160> 6
<170> PatentIn version 3.3
<210> 1
<211> 987
<212> DNA
<213> 基因NtBRC1的核苷酸序列
<400> 1
atgtatccgc caagcaacag ctgcaactac agccccattt tcaacatccc ttctccttgt 60
atgcaatatg gagacgaact attcttccaa tattatcctg accatttcct tcaacagcaa 120
caagtgcctt tgatagaaga tcagagtgtt gacatcttag ctgattgcac tgagaatgtt 180
actaacgaag aaactgtcat caatactgat actgtaaaag ttctttatga cacaggagct 240
gttacaaaca gtcagtgttg gggaggaaat gaagaagtag aagaaggccg cgaaaacaaa 300
agaaatgaca tgagaagcac cattagtatt attcatgtac ggaaaaacaa gaaatgttcc 360
aataaagatc gacatagcaa gattaacact gctcgtggcc tcagagaccg aaggatgaga 420
ctttcccttg atgcagctcg caagtttttc agtttacaag acatgttggg gtccgataag 480
gcaagtaaaa ctgtagaatg gttgcttatc aaatcggggt ctgaaatcga agagctagcc 540
aaaggcaata aaggaggagg cattcctaaa caaagctgca gtactactaa tggaattggt 600
gcaattagta ctgcaatatc ctctatttct gagtgtgagg ttatatcagg aactgatgaa 660
tctttctcta ttacttataa aaagaagctg aaaactgcta aaggagcctc gaaaaagacg 720
gctaaaactg ctcgtagagc tgcatttgat cgtcttatta caagggaaac gaggaatcaa 780
gcaagggcta gggctagaga gagaacaaaa ataaagaaaa gcctcggtaa atccaaagag 840
aacagtgctg attactgtaa tttggtggat aattatggag attggagtca atttagtatc 900
ttcaactatc agaaaaatgc agttggaatt tcccatgatc aggtgggttc aataattaaa 960
caacatgatt ttttaggatt tcaatag 987
<210> 2
<211> 328
<212> PRT
<213> 基因NtBRC1编码的氨基酸序列
<400> 2
Met Tyr Pro Pro Ser Asn Ser Cys Asn Tyr Ser Pro Ile Phe Asn Ile
1 5 10 15
Pro Ser Pro Cys Met Gln Tyr Gly Asp Glu Leu Phe Phe Gln Tyr Tyr
20 25 30
Pro Asp His Phe Leu Gln Gln Gln Gln Val Pro Leu Ile Glu Asp Gln
35 40 45
Ser Val Asp Ile Leu Ala Asp Cys Thr Glu Asn Val Thr Asn Glu Glu
50 55 60
Thr Val Ile Asn Thr Asp Thr Val Lys Val Leu Tyr Asp Thr Gly Ala
65 70 75 80
Val Thr Asn Ser Gln Cys Trp Gly Gly Asn Glu Glu Val Glu Glu Gly
85 90 95
Arg Glu Asn Lys Arg Asn Asp Met Arg Ser Thr Ile Ser Ile Ile His
100 105 110
Val Arg Lys Asn Lys Lys Cys Ser Asn Lys Asp Arg His Ser Lys Ile
115 120 125
Asn Thr Ala Arg Gly Leu Arg Asp Arg Arg Met Arg Leu Ser Leu Asp
130 135 140
Ala Ala Arg Lys Phe Phe Ser Leu Gln Asp Met Leu Gly Ser Asp Lys
145 150 155 160
Ala Ser Lys Thr Val Glu Trp Leu Leu Ile Lys Ser Gly Ser Glu Ile
165 170 175
Glu Glu Leu Ala Lys Gly Asn Lys Gly Gly Gly Ile Pro Lys Gln Ser
180 185 190
Cys Ser Thr Thr Asn Gly Ile Gly Ala Ile Ser Thr Ala Ile Ser Ser
195 200 205
Ile Ser Glu Cys Glu Val Ile Ser Gly Thr Asp Glu Ser Phe Ser Ile
210 215 220
Thr Tyr Lys Lys Lys Leu Lys Thr Ala Lys Gly Ala Ser Lys Lys Thr
225 230 235 240
Ala Lys Thr Ala Arg Arg Ala Ala Phe Asp Arg Leu Ile Thr Arg Glu
245 250 255
Thr Arg Asn Gln Ala Arg Ala Arg Ala Arg Glu Arg Thr Lys Ile Lys
260 265 270
Lys Ser Leu Gly Lys Ser Lys Glu Asn Ser Ala Asp Tyr Cys Asn Leu
275 280 285
Val Asp Asn Tyr Gly Asp Trp Ser Gln Phe Ser Ile Phe Asn Tyr Gln
290 295 300
Lys Asn Ala Val Gly Ile Ser His Asp Gln Val Gly Ser Ile Ile Lys
305 310 315 320
Gln His Asp Phe Leu Gly Phe Gln
325
<210> 3
<211> 20
<212> DNA
<213> 正向引物
<400> 3
atgtatccgc caagcaacag 20
<210> 4
<211> 21
<212> DNA
<213> 反向引物
<400> 4
gctattgaaa tcctaaaaaa t 21
<210> 5
<211> 26
<212> DNA
<213> NtBRC1-OE-F
<400> 5
ggatccatgt atccgccaag caacag 26
<210> 6
<211> 26
<212> DNA
<213> NtBRC1- OE-R
<400> 6
ctcgagctat tgaaatccta aaaaat 26
- 还没有人留言评论。精彩留言会获得点赞!
- 一种烟草腋芽生长调控基因NtMOC1及其克隆方法与应用与流程
- 一种烟草KC1基因及其制备方法和应用与流程
- 一种烟草AKT1基因及其制备方法和应用与流程
- RPS23基因作为内参基因在定量检测螟黄赤眼蜂基因表达量中的应用的制造方法与工艺
- 一种烟草抗旱基因NtSAP5及其克隆方法与应用与流程
- 一种烟草HKT1基因及其制备方法和应用与流程
- PABPC1基因作为内参基因在定量检测稻螟赤眼蜂基因表达量中的应用的制造方法与工艺
- 一种应用内参基因快速检测肉制品中羊源性成分含量的方法与流程
- 用于甜瓜果实基因PCR表达分析的内参基因及该内参基因的稳定性验证方法与流程
- 一种烟草干旱响应基因NtXERICO及其克隆方法与应用与制造工艺