一种螺旋度可调节的多肽化合物的合成方法与流程
本发明属于多肽结构稳定和修饰技术领域。更具体地说,本发明涉及一种通过构建手性侧链来稳定和调节多肽alpha螺旋,并从而调节多肽穿膜性、与靶点的亲和性和血清稳定性的方法以及由该方法构建的螺旋度可调节的多肽化合物。
背景技术:
研究表明,在蛋白蛋白相互作用中,参与结合作用的往往只有很小的一段多肽序列,而其它部分是为了维持蛋白特定构象而存在。alpha螺旋构象作为蛋白最重要的二级构象之一,在细胞生理过程中起着至关重要的作用,超过60%的蛋白-蛋白相互作用的区域为alpha螺旋构象。因此,具有特定alpha螺旋构象的多肽在靶向特定蛋白蛋白相互作用过程中,有非常重要的作用和药物前景,这也是目前多肽药物关注的方向。此外,使多肽具有稳定的alpha螺旋构象还能大幅提升多肽的抗蛋白酶降解能力和穿膜能力,从而提高多肽的成药性。
为了使多肽具有稳定的alpha螺旋构象,研究人员开发了各种各样的多肽构象稳定技术,如盐桥、金属螯合、HBS、构建共价侧链等。其中构建共价侧链是其中较为常用的方法,最早出现的是以酰胺键的形成作为侧链构建方法。此后二硫键,碳碳双键的形成等均被用于构建侧链以稳定多肽的alpha构象。
在侧链共价键的构建过程中偶联的侧链一般在alpha螺旋的同一面上,所以侧链偶联氨基酸的位置一般为i/i+3,i/i+4,i/i+7,i/i+11等。其中应用较多的是i/i+4与i/i+7。
以上介绍的方法都有一定的局限性,例如酰胺键构建的环肽由于易水解而使成药性较差;以二硫键形成的环肽受到二硫键本身的稳定性影响,其应用也很有限;基于叠氮与炔反应的侧链形成的三氮唑本身就是一个药效基团,其对多肽成药性影响可能更大;以烯烃复分解反应构建的环肽是目前应用最广成药性最好的多肽稳定方法,但是,重金属催化剂的引入导致多肽分离纯化成本增加,使其在大规模生产中受限。
因此,发展一种合成简单,绿色,高效的多肽螺旋稳定方法,同时,在多肽螺旋稳定的基础上发展螺旋度可调节的多肽化合物,对于促进新型多肽药物的研发提高多肽的成药性具有重要的意义。
技术实现要素:
本发明的目的是提供一种通过构建新型的含有2位碳手性的侧链来稳定多肽的alpha螺旋的,更重要的是通过改变侧链手性基团的大小可以调节短肽的alpha螺旋二级构象,并且这种带有侧链手性修饰的多肽化合物的穿膜性、与靶点的亲和性和血清稳定性都有明显提高。
进而,本发明提供了一种螺旋度可调节的多肽化合物的合成方法,该合成方法具体包括如下步骤:
(ⅰ)将多肽的羧基端连接非天然氨基酸,所述非天然氨基酸具有如下结构式:
其中R1为甲基、苯基、乙基、异丙基、叔丁基、苄基、羟基或羧基,R2为氢或甲基;
(ⅱ)将步骤(i)所得的产物经过巯基-烯反应获得2位碳手性修饰的多肽化合物,该侧链偶联氨基酸的位置为i/i+4;
(ⅲ)将步骤(ⅱ)所得的2位碳手性修饰的多肽化合物在树脂上进行关环;
(ⅳ)使用剪切液将步骤(ⅲ)所得的产物从树脂上剪切下来,经过分离纯化获得目标产物;
该目标产物的结构式如下:
其中,R1和R6各自独立地为氢或甲基,R2为甲基、苯基、乙基、异丙基、叔丁基、苄基、羟基或羧基,R3、R4、R5各自独立地为氨基酸残基,侧链碳中心的构型为R。
本发明中,所述步骤(ⅱ)中的巯基-烯反应是先将步骤(i)的产物与半胱氨酸或半胱氨酸衍生物进行光聚合反应,然后通过酰胺键的形成合成硫醚侧链修饰的多肽化合物。
本发明中,所述步骤(ⅱ)和步骤(ⅲ)的反应式如下:
本发明中,化合物1与半胱氨酸(Fmoc-Cys-OH)及其衍生物的摩尔比为1:10。
本发明中,化合物2、PyBOP、HOBt、NMM的摩尔比为1:2.4:2.4:4。
本发明中,步骤(ⅳ)中使用的剪切液为体积比为9.5:0.25:0.25的三氟乙酸/三异丙基硅烷/水溶液。
本发明提出了一种螺旋度可调节的多肽化合物合成方法,通过构建新型的侧链不但能稳定多肽alpha螺旋的方法,而且该方法通过调节侧链碳手性基团的大小可以控制alpha螺旋的含量。通过该方法合成的生物活性多肽,能显出良好的螺旋形,稳定性以及细胞穿透能力。该方法通过调节手性基团的大小来调节多肽螺旋度,穿膜性,靶点亲和力等是目前本领域的其他稳定方法所不具备的。
附图说明
图1为实施例2中R2=methyl时Ac-cyclo(1,5)-CAAAS5(2-CH3)-NH2(1a/b)在水中的CD图。
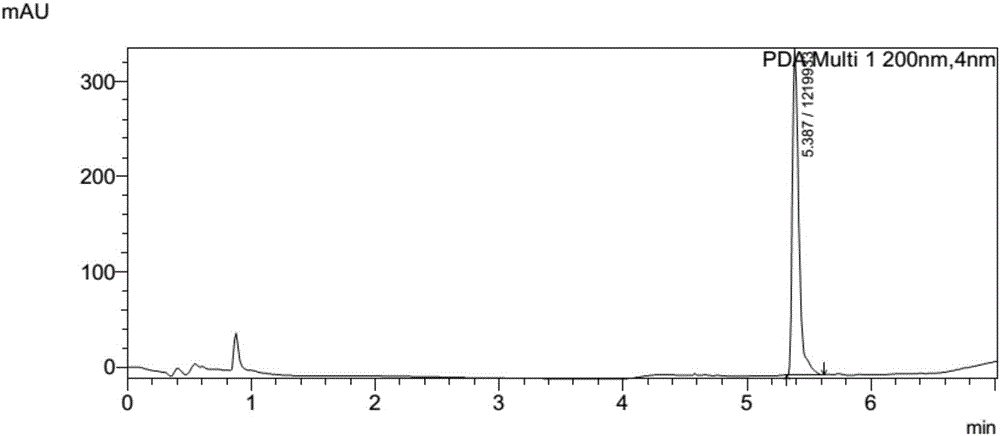
图2为实施例2中多肽1a Ac-(cyclo-1,5)-[CysAlaAlaAlaS5(2-Me)(S)]-NH2的LC-MS检测的LC图。
图3为实施例2中多肽1a Ac-(cyclo-1,5)-[CysAlaAlaAlaS5(2-Me)(S)]-NH2的LC-MS检测的MS图。
图4为实施例2中多肽1b Ac-(cyclo-1,5)-[CysAlaAlaAlaS5(2-Me)(R)]-NH2的LC-MS检测的LC图。
图5为实施例2中多肽1b Ac-(cyclo-1,5)-[CysAlaAlaAlaS5(2-Me)(R)]-NH2的LC-MS检测的MS图。
图6为实施例1中不同序列的R型非对映异构体Ac-cyclo(1,5)-CAXAS5(2-methyl)-NH2(2b-10b)在水中的的CD图。
图7为实施例1中不同序列的S型非对映异构体Ac-cyclo(1,5)-CAXAS5(2-methyl)-NH2(2a-10a)在水中的的CD图。
图8为实施例3中多肽1b的热稳定CD图谱和盐酸胍稳定性以及胰蛋白酶降解实验结果,其中图A为热稳定性CD图谱,图B为盐酸胍稳定性以及热稳定性统计结果图,图C为血清稳定性图。
图9为实施例4中生物活性多肽ER-1a/b以及PDI-1/2-a/b在20%TFE/H2O中的CD结果。
图10为实施例4中生物活性多肽ER-1a/b以及PDI-1/2-a/b与ERα和MDM2的结合力测定实验。
图11为实施例5中多肽11-a/b以及12a/b与HEK293T细胞孵育后的穿膜性测定实验和CD图,其中图A为穿膜性测定实验图,图B为CD图。
图12为实施例5中多肽11a FITC-(cyclo-2,6)-[βAlaCysArgAlaArgS5(2-Ph)(S)]-NH2的LC-MS检测的LC图。
图13为实施例5中多肽11a FITC-(cyclo-2,6)-[βAlaCysArgAlaArgS5(2-Ph)(S)]-NH2的LC-MS检测的MS图。
图14为实施例5中多肽11b FITC-(cyclo-2,6)-[βAlaCysArgAlaArgS5(2-Ph)(R)]-NH2的LC-MS检测图,其中图A为LC图,图B为MS图。
图15为实施例5中多肽12a FITC-(cyclo-2,6)-[βAlaCysArgArgArgS5(2-Ph)(S)]-NH2的LC-MS检测的LC图。
图16为实施例5中多肽12a FITC-(cyclo-2,6)-[βAlaCysArgArgArgS5(2-Ph)(S)]-NH2的LC-MS检测的MS图。
图17为实施例5中多肽12b FITC-(cyclo-2,6)-[βAlaCysArgArgArgS5(2-Ph)(S)]-NH2的LC-MS检测的LC图。
图18为实施例5中多肽12b FITC-(cyclo-2,6)-[βAlaCysArgArgArgS5(2-Ph)(S)]-NH2的LC-MS检测的MS图。
具体实施方式
研究表明,相对于小分子药物,多肽分子具有大接触面积,低毒性和低免疫原性等特点,使其可以靶向许多小分子药物无法成药的靶点。但是,在溶液中的多肽往往不具有稳定的二级构象。如何使少于20个氨基酸的多肽在水相中具有稳定构象一直是多肽化学中的研究热点。alpha螺旋是蛋白中最重要的二级结构之一,在很多重要的生理过程,如信号传导、细胞凋亡中等起着重要作用,所以如果能够使多肽具有稳定的alpha螺旋结构将会使这些过程更加容易调控。以构建共价键的侧链来稳定多肽alpha螺旋构象是其中应用最多的策略之一。
本发明人以五肽为模型,通过构建一种新的侧链使得多肽alpha螺旋的稳定性有明显提高,而且这一侧链具有很高的序列耐受能力,更重要的是通过调节侧链碳手性基团的大小可以控制alpha螺旋的含量。同时,这一方法被应用于具有生物活性的靶点多肽,通过该方法稳定的多肽表现出良好的细胞穿透性、与靶点结合的亲和力以及稳定性,对于多肽药物研发,提高多肽的成药性有非常重要的意义和价值。首先,本发明人运用thiolene反应构建2位碳手性侧链稳定的多肽化合物,并进一步将多肽环化。之后通过CD证明了这一碳手性中心对稳定多肽的alpha螺旋二级结构效果良好,而且对多肽序列有良好的耐受性。上述侧链在多肽化合物中的位置有如下结构通式:
其中,R1和R6各自独立地为氢或甲基,R2为甲基或苯基以及其他基团,R3~R5各自独立地为氨基酸残基侧链,侧链碳中心的构型为R。
制备上述侧链稳定的多肽化合物的核心反应步骤如下:
化合物1是将多肽的羧基端连接非天然氨基酸。其中的天然氨基酸是由Fmoc(芴甲氧羰基)保护的固相接肽完成,所述非天然氨基酸(简称为S5[2-R2])的结构式如下:
所述非天然氨基酸可通过固相接肽连接到树脂上,其中R1为甲基/苯基或其他基团,R2为氢或甲基。
化合物2,是由化合物1与半胱氨酸(Fmoc-Cys-OH)及其衍生物进行反应而得。反应条件设定为:取化合物1 0.5mmol,加入光引发剂MAP:4-methoxyacetophenone,MMP:2-hydroxy-4’-(2-hydroxyethoxy)-2-methylpropiophenone各1.0mmol,Fmoc-Cys-OH衍生物5.0mmol,然后加入20ml DMF作溶剂,Ar保护于365nm紫外光下反应3h,滤掉反应液洗涤即可得化合物2。
将上一步得到的化合物2加入到10ml DMF中,加入PyBOP,HOBt,NMM,在N2鼓泡的条件下反应4h,滤掉反应液,用DMF和DCM交替洗涤3次,接下来用25%的吗啡啉脱除Fmoc-保护基,即得到关环的多肽树脂3。
多肽树脂3进一步乙酰化,把树脂抽干,加入10ml三氟乙酸(TFA)/三异丙基硅烷(TIS)/水(H2O)(9.5:0.25:0.25体积/体积)的剪切液中反应3h,用氮气将剪切液吹干,10ml乙醚/正己烷(4:1)沉淀;HPLC纯化,冻干,得到干净的化合物3,即2’碳手性侧链修饰的多肽化合物。
HPLC纯化得化合物3,将化合物3溶于4ml水中。即2’碳手性侧链修饰的多肽化合物。化合物3含有两个异构体,其中出峰在前的称为化合物3A(S),出峰在后的为化合物3B(R)。
将得到的化合物溶于水中测CD谱。化合物3B具有良好的alpha螺旋,化合物3A主要表现出无规卷曲的构象。
本发明人进而通过CD结果证实了这一侧链碳手性对稳定多肽的alpha螺旋二级结构效果很好,而且对多肽序列有良好的耐受性。并且后续的多肽穿膜性实验、与靶点的结合力实验以及血清稳定性实验都表明这一侧链碳手性对于提高多肽穿膜性实验、结合力和稳定性方面也有重要的作用。
综上所述,本发明提供了一种通过构建新型的侧链稳定多肽alpha螺旋的方法,而且该方法在一定范围内可通过调节侧链碳手性基团的大小来控制alpha螺旋的含量。通过该方法稳定的生物活性多肽,便显出良好的螺旋形,稳定性以及细胞穿透能力。该方法通过调节手性基团的大小来调节多肽螺旋度,穿膜性,靶点亲和力等是目前本领域的其他稳定方法所不具备的。
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件中所述的条件,或按照制造厂商所建议的条件。
除非另行定义,文中所使用的所有专业与科学用语与本领域熟练人员所熟悉的意义相同。此外,任何与所记载内容相似或均等的方法及材料皆可应用于本发明中。文中所述的较佳实施方法与材料仅作示范之用。
实施例1 R2=Phenyl时2位碳手性侧链稳定的多肽Ac-cyclo(1,5)-CAAAS5(2-phenyl)-NH2的合成:
S5(2-phenyl)的结构式为:
首先是以Fmoc固相多肽合成法合成NH2-AAAS5(2-phenyl)-MBHA树脂:
具体路线如下:
具体操作为:
1.接第一个氨基酸:称取1.0g MBHA树脂于100ml接肽管中,加入20ml N-甲基吡咯烷酮(NMP)鼓氮气溶胀30min;滤掉溶剂加入体积比为25%吗啡啉的NMP溶液,鼓氮气30min,洗涤;连接反应:加入Fmoc-S5(2-phenyl)-OH(0.4M in NMP)溶液,HCUT(0.38M in NMP),DIEA按5.0ml/5.0ml/0.71ml混匀加入树脂中鼓氮气120min,滤掉反应液。洗涤:将接肽管中的溶剂抽干,将树脂用NMP(10ml*3)洗涤三次,每次一分钟;
2.接第二个氨基酸:脱保护:加入体积比为25%吗啡啉的NMP溶液,鼓氮气30min,洗涤;连接反应:将配制好的Fmoc-Ala-OH(0.4M in NMP)溶液,6-氯苯并三氮唑-1,1,3,3-四甲基脲六氟磷酸酯(HCUT)(0.38M in NMP),DIEA按7.5ml/7.5ml/1ml混匀加入树脂中鼓氮气50min;滤掉反应液,洗涤然后进行下一步操作。
3.接第三个氨基酸:操作同2接第三个Ala。
4.接第四个氨基酸:操作同2接第四个Ala。脱保护:加入体积比为25%吗啡啉的NMP溶液,鼓氮气30min,洗涤
5.滤掉反应液,将树脂依次用NMP(10ml),二氯甲烷(DCM)(10ml),甲醇(MeOH)(10ml)交替洗涤,抽干保存或用于下一步反应。
通过thiol-ene反应(巯基-烯反应)来完成侧链构建。
首先进行光聚合反应:
所获得反应中间体的结构式为:
具体操作为:称取1.0g H2N-AAAS5(2-phenyl)-MBHA树脂于100ml烧瓶中,依次加入1.4g Fmoc-Cys-OH,330mg DMPA,50ml DMF;用氩气换气三次除掉溶剂中的氧;将该烧瓶置于光反应器中搅拌下反应3h;然后将反应树脂转入接肽管中,滤除反应液,用DMF(10ml),DCM(10ml),交替洗涤,抽干得H2N-AAAS5(2-phenyl)(Fmoc-Cys-COOH)-MBHA树脂。
取30mg H2N-AAAS5(2-phenyl)(Cys-COOH)-MBHA溶胀,加入2.4equiv的PyBOP,HOBt,4equiv NMP在氮气鼓吹下反应4h。然后使用morphine脱除Fmoc-保护基,加入1ml Ac2O:DIEA:NMP=85:315:1600乙酰化试剂,在氮气鼓吹下反应1h,即得到多肽末端氨基乙酰化的多肽。
使用剪切液(TFA:TIS:H2O=95:2.5:2.5)把多肽从树脂上剪切下来,滤掉树脂,用N2把剪切液吹掉,用冷却的(乙醚:正己烷=1:1)沉淀,沉淀加水和乙腈溶解后用HPLC纯化,460nm*2.5mm C18反相色谱,A液:0.1%TFA/H2O,B液:0.1%TFA/乙腈;溶剂梯度:0-10min 5-15%;10-30min 15-55%;Rt=26.000min。得到HPLC色谱图及LCMS检测图。
产物冻干后取2mg溶于0.5ml氘代DMSO中核磁检测:
1H NMR(400MHz,DMSO)δ8.26(d,J=7.8Hz,1H),8.29(d,J=6.5Hz,1H),8.19(d,J=8.0Hz,1H),7.96(d,J=8.3Hz,1H),7.15(d,J=7.1Hz,1H),7.23-7.09(m,5H),6.98(s,1H),6.84(s,1H),4.21–4.10(m,5H),2.96(dd,J=15.3,5.7Hz,1H),2.76–2.71(m,1H),1.83(s,3H),1.82(t,J=5.9,3H),1.80–1.21(m,17H),1.09(m,1H).
将Ac-cyclo(1,5)-CAAA S5(2-phenyl)-NH2两种异构体(S-phenyl)/(R-phenyl)溶于10mMPBS溶液中测定CD图谱:取0.1-0.5mg样品分别溶于0.5ml 10mmol PBS中测CD波长为190nm-250nm处的色谱图,然后根据以下公式计算得alpha螺旋含量。
alpha螺旋计算公式:
fH=([θ]obs215-[θ]C)/([θ]∞215-[θ]C)
[θ]∞215=(-44000+250T)(1-к/Np)
[θ]C=2220-53T=1054
T=22℃,к=4.0,Np=6(对于五肽),C为样品摩尔浓度M。
从CD图的差别中可以看出两种异构体只有在HPLC分离中出峰靠后的异构体B具有稳定的alpha螺旋。
以2b为参照,计算得到其他短肽的相对螺旋度如下表所示:
由此可知,改变五肽的侧链2’带有手性的氨基酸的种类,如S5(2-(R)phenyl)变成S5(2-(R)methyl)不利于alpha螺旋。这就表明侧链手性基团的大小会对短肽的alpha螺旋产生至关重要的作用。因此本方法也可已通过改变侧链手性基团的大小来调节短肽的alpha螺旋二级构象。
实施例2R2=CH3时2’手性碳中心侧链稳定的多肽的合成及CD图谱的测定
将S5(2-phenyl)换成手性基团较小的S5(2-methyl)用同样的合成路线,得到了两个非对映异构体,同样根据HPLC上出峰时间的先后命名为Ac-cyclo(1,5)-CAAA S5(2-(S)methyl)-NH2-AMSA和Ac-cyclo(1,5)-CAAAS5(2-(R)methyl)-NH2,LCMS鉴定,及核磁氢谱。
1H NMR(400MHz,DMSO)δ8.62(d,J=7.5Hz,1H),8.18(d,J=7.7Hz,1H),8.14(d,J=6.9Hz,1H),7.73(d,J=8.4Hz,1H),7.17(d,J=7.1Hz,1H),7.06(s,1H),6.93(s,1H),4.25–4.13(m,5H),2.94(dd,J=12.3,4.5Hz,1H),2.81–2.75(m,1H),1.83(s,3H),1.65(m,1H),1.61-1.02(m,17H),0.84(m,1H),0.79(d,J=4.04,3H)。
测定Ac-cyclo(1,5)-CAAAS5(2-methyl)-NH2A/B的CD(图1),同样只有B异构体才有alpha螺旋稳定效果。
实施例3多肽1b的热稳定性,盐酸胍稳定性测试和血清稳定性测定。
用本发明介绍的同实施例1相同的方法合成多肽1b,将多肽1b在不同温度下以水为溶剂利用CD测定二级结构,结果表明,在不同温度下,2位手性基团诱导的多肽1b都保持着良好的而稳定的α-螺旋二级结构。
将定量的多肽样品平行6份溶解在水中,在37℃条件下与800微升人血清混合孵育,分别在0h,0.5h,1h,2h,4h,8h时,加入蛋白沉淀剂(乙腈:水=3:1)沉淀,离心去除血清蛋白。然后立刻用LC-MS定量分析多肽剩余的量。结果表明,经过2位手性基团诱导产生的具有良好α-螺旋的多肽具有良好的血清稳定性,在血清中孵育8h后,依然有超过60%剩余。
盐酸胍稳定性实验结果:
将多肽与不同浓度的盐酸胍(0-8M)孵育1小时,测量多肽的二级结构的变化。结果如图8B所示,随着盐酸胍浓度的增加,多肽螺旋度略有下降,但是在8M的盐酸胍条件下,仍有80%左右的保持。
实施例4ER-1a/b、ER-2a/b和PDI-1a/b、PDI-2a/b与靶点结合力的测定
用本发明介绍的同实施例1同样的合成方法合成多肽ER-1a/b、ER-2a/b和PDI-1a/b、PDI-2a/b,如下表所示:
多肽ER-1a/b、ER-2a/b和PDI-1a/b、PDI-2a/b的序列及分子量
分别作用于已知的药物靶点ER-α和MDM2。利用荧光共振实验在96孔板上测定与蛋白的结合力。在96孔板上将10nM连有FITC荧光基团的多肽ER-1a/b、ER-2a/b分别加入孔中,孔中加有不同浓度梯度的纯化好的ER-α蛋白,在缓冲液(10μM的17-β雌二醇、20mM的Tris-HCl PH 8.0、25mM NaCl、10%甘油、10μMβ雌二醇、1mM的TCEP(3(2-羧乙基)膦))中混合,在4℃下暗处孵育1h,之后在16℃下检测荧光(吸收波长485nm,发射波长520nm),最后用origin 9.0处理数据得出ER-1a/b、ER-2a/b对ER-α蛋白的结合力曲线。(图7)
同理PDI-1a/b、PDI-2a/b对MDM2的结合力曲线也如上所述得到。只是缓冲液更换为:140mM NaCl,50mM,Tris pH 8.0。
结果表明B型异构体(R性)具有良好的与靶点蛋白结合的能力。
实施例5多肽11a/b的穿膜性测定
用本发明介绍的同实施例1同样的合成方法合成多肽11a/b和12a/b:
FITC-(cyclo-2,6)-[βAlaCysArgAlaArgS5(2-Ph)(S)]-NH2、
FITC-(cyclo-2,6)-[βAlaCysArgAlaArgS5(2-Ph)(R)]-NH2和
FITC-(cyclo-2,6)-[βAlaCysArgArgArgS5(2-Ph)(S)]-NH2、
FITC-(cyclo-2,6)-[βAlaCysArgArgArgS5(2-Ph)(R)]-NH2
将这四条经HPLC纯化后的多肽用DMSO溶解(浓度1mM),与经DMEM(10%FBS)培养的HEK293T细胞(提前一天传代,约占80%)混合,使多肽最终浓度为5μM。然后在37℃下孵育1h,然后用PBS洗涤细胞3次,用4%的多聚甲醛固定10min后,再用PBS洗涤3次,然后用1μg/ml的DAPI(4',6-二脒基-2-苯基吲哚)染色5min后,用共聚焦显荧光微镜观察多肽穿膜情况。结果表明B型异构体(R性)具有良好的细胞穿透性。
多肽1-12a/b的计算分子量以及LC/MS观测到的分子量
本发明所涉及的多个方面已做如上阐述。然而,应理解的是,在不偏离本发明精神之前提下,本领域专业人员可对其进行等同改变和修饰,所述改变和修饰同样落入本申请所附权利要求的覆盖范围。
- 还没有人留言评论。精彩留言会获得点赞!