一种高纯度环己肽类化合物的制备方法与流程
技术领域:
本发明属于工业微生物
技术领域:
,具体涉及一种高纯度的环己肽类化合物FR179642的制备方法。
背景技术:
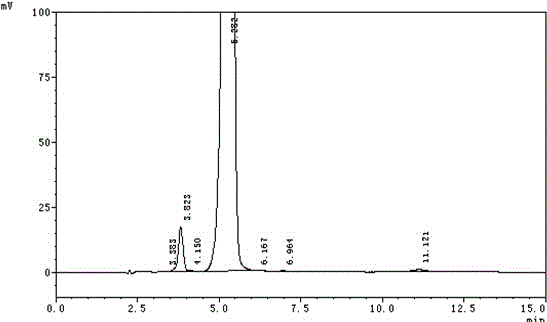
:真菌对于健康人体而言通常是条件致病菌,当肌体免疫力下降,外部因素不良时,就可能造成局部或全身真菌感染。近年来,随着广谱抗生素免疫抑制剂和糖皮质激素的不当应用以及器官移植、放疗化疗、外科介入等的广泛应用,导致深部真菌感染发病率日益增高。真菌感染会侵犯内脏器官和深部组织,因此,有效控制深部真菌病具有十分重要的临床意义。棘白菌素类是近年来新上市的一种抗真菌药物,能抑制真菌细胞壁的β-1,3-D-葡萄糖合成酶,由于哺乳动物细胞缺乏β-1,3-D-葡萄糖合成酶,故该类化合物对真菌细胞具有高度的特异性,能迅速杀灭真菌而对人体正常细胞影响较小,因而疗效好,安全性高。目前已上市的此类药物有:卡泊芬净、米卡芬净和阿尼芬净。米卡芬净(Micafungin,其结构式如式1所示)由日本藤泽公司开发,于2002年12月在日本上市,商品名为Fungusrd,2005年3月通过美国FDA认证,目前被批准用于治疗食道念珠菌感染、骨髓移植及ADS患者中性粒细胞减少症的预防治疗。米卡芬净由环己肽类化合物FR179642(结构式如式2所示)修饰而得到的水溶性半合成的新型抗真菌药,环己肽类化合物FR179642是鞘茎点霉Coleophomaempetri发酵代谢产物。因此,要得到高纯度的米卡芬净,纯度高的式2即环己肽类化合物FR179642是关键。式1:米卡芬净式2:FR179642目前,关于环己肽类化合物FR179642提取分离的文献报道较少。中国专利CN102443050A披露了以一种环脂肽化合物为前体通过酶解掉侧链得到FR179642,并报道了该种前体环脂肽化合物的分离提纯方法,但没有涉及到FR179642的提纯方法。此外还有文献中报道由FR179642制备米卡芬净的合成方法(Ueda,S.;Ezaki,M.;Tanaka,M.;etal.StudiesonanovellipopeptideacylasefromStreptomycesspp.forproductionofFR179642,akeyintermediateofantifungallipopeptidedrugFK463.38thIntersciConfAntimicrobAgentsChemother(Sept241998,SanDiego)1998,AbstF-145)。中国专利CN201110244301.5公开了一种纯化环己肽类化合物FR179642的方法:将发酵液过滤后进行树脂吸附、解吸、浓缩、结晶后得到环己肽类化合物FR179642粗品,然后再借助反向填料C18对粗提物进行层析分离,最后得到较高纯度(98.1%~98.6%)的环己肽类化合物FR179642。与本发明技术方案相比,现有技术有如下缺点:1)进行树脂吸附后再进行反向填料层析分离,增加了操作步骤和生产成本;2)借助反向填料C18,价格较贵;3)解吸、结晶、层析分离过程中均使用了甲醇这种有毒溶剂,对操作人员的身体健康构成威胁,且对环境造成污染,不适合工业化大规模生产。因此,本领域急需找到一种操作步骤简单、减小生产成本的纯化方法,克服上述现有技术中存在的缺陷,并且,同时能进一步提高环己肽类化合物FR179642产品的纯度。技术实现要素:本发明的目的在于开发一种操作简便、所用溶剂毒性低、适合于工业生产的环己肽类化合物FR179642的制备方法,其特征在于采用大孔树脂提取分离FR179642,工艺简单易行,适宜于工业化生产;并通过低温重结晶进一步纯化,以制备高纯度的环己肽类化合物FR179642产品。本发明提供了一种环己肽类化合物FR179642的纯化方法,所述的方法包括如下步骤:1)向环己肽类化合物FR179642的发酵液中加入助滤剂,搅拌均匀;2)将步骤1)中加入助滤剂的发酵液进行过滤或者离心,得到环己肽类化合物FR179642的滤液,并将滤液浓缩;3)将步骤2)中的浓缩液导入大孔吸附树脂进行富集;4)将极性溶剂与水混合而成的溶液作为解吸液对上述大孔吸附树脂进行解吸并收集,得到解吸混合液;5)向解吸混合液中加入脱色物质,搅拌均匀、过滤,得到脱色液;6)将脱色液浓缩,结晶,真空加热干燥,得到环己肽类化合物FR179642的精粉。其中,步骤1)中的助滤剂为珍珠岩或硅藻土,其用量为发酵液的2~6%(w/v)。步骤2)中的固液分离方式为离心或板框过滤。步骤2)和步骤6)中的浓缩方法为纳滤、旋蒸或闪蒸中的一种或多种,步骤2)浓缩至浓缩液中环己肽类化合物FR179642含量为1mg/ml~10mg/ml,步骤6)浓缩至浓缩液中环己肽类化合物FR179642含量为10mg/ml~50mg/ml。步骤3)中的大孔吸附树脂是非极性或中等极性吸附树脂。步骤3)中的大孔吸附树脂选自XAD1600、XAD16HP、XAD1180、XAD7HP、XAD18、XAD16N或XAD761,树脂柱径高比为1/5~1/10,其用量与纳滤浓缩液中环己肽类化合物FR179642粗品的体积重量比为70~150:1(ml/g),吸附流速为0.8~3.2BV/h。步骤4)在解吸前,需用纯化水水洗大孔吸附树脂3~7BV,流速1~2BV;解吸极性溶剂为异丙醇、乙醇、丙酮中的一种,含量为2%~15%(v/v),体积为2~10BV,流速为0.5~2.0BV;解吸后,收集产品含量在500μg/ml,纯度在90%以上的组分,组成解吸混合液。步骤5)中的脱色物质为767针用活性炭,加入量为0.05~0.15%(w/v),优选0.08~0.10%;搅拌脱色时间为10~60min,优选20~30min。步骤6)中的结晶温度控制在0~25℃,优选10~15℃;结晶时间为2~16h,优选8~12h;真空加热干燥温度控制在25~30℃,干燥时间为4~16h。步骤6)中结晶可以为一次结晶或重复多次结晶,重复结晶时可采用纯化水作为溶剂溶解前次结晶品,用量为每克湿晶体需加入纯化水0.5~5ml。环己肽类化合物FR179642是合成新型抗真菌药米卡芬净的关键中间体,本发明所得的FR179642产品的含量为97%(重量)以上,纯度高达99%以上,样品总的回收率大于60%。相对于现有技术,本发明的主要优点在于:1、本发明提供了一种成本低廉的纯化环己肽类化合物,特别是棘白菌素类化合物FR179642的新方法;2、本发明采用大孔吸附树脂结合使用脱色剂和低温重结晶的方法对环己肽类化合物FR179642进行纯化,具有纯化路线短、纯化条件温和、产品收率高、操作简便等优点,在较大程度上减轻了工艺操作和对设备的要求,降低了生产成本;3、本发明在采用低浓度的极性有机溶剂(2%~15%)做洗脱剂,洗脱效率高,大大提高产品的收率,洗脱液用量较少(洗脱液体积为2~10倍浓缩液体积),对环境污染小,对操作人员危害较小;4、本发明后处理过程加入脱色剂对产品进行处理,可显著提高产品色泽和纯度;5、本发明提供的方法制备的环己肽类化合物FR179642精粉较稳定,纯度高,有利于终产品的质量控制,适宜于工业化生产;6、使用本发明提供的方法制备的目标产物FR179642完全可以满足合成新型抗真菌药米卡芬净要求,便于米卡芬净规模化合成。附图说明图1为实施例1中FR179642发酵液滤液的HPLC图谱。图2为实施例1中FR179642一次结晶潮品的HPLC图谱。图3为实施例1中FR179642二次结晶精粉的HPLC图谱。具体实施例下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不意味着对本发明有任何限制。本发明所用的环己肽类化合物FR179642发酵液为重庆乾泰生物医药有限公司发酵部门用微生物培育手段,得到的FR179642发酵液或者现有技术中公开的方法制备得到含有FR179642的发酵液,包括但不限于CN102443050A、CN201110244301.5公开的方法得到的FR179642发酵液;XAD1600树脂、XAD1180树脂均来自于郑州勤实科技有限公司;乙醇、丙酮等溶剂均为市售。本发明所用的高效液相色谱仪为岛津公司。下述实施例中环己肽类化合物FR179642HPLC检测所用的方法:反相HPLC分析采用的色谱柱为C18柱(4.6mm*250mm*5um),保持在40℃,以0.05mol/LNaH2PO4:乙腈=97:3(v/v)作流动相,流速为1.0ml/min,并在210nm下UV检测。具体实施例实施例1含有FR179642发酵液22.7L,发酵单位为700μg/ml。向其中加入1kg硅藻土,搅拌半小时后离心分离,得到18.9L滤液(HPLC的图谱见图1和表1)。将滤液进行闪蒸浓缩,得到浓缩液3.8L,单位为3642μg/ml。将浓缩液导入XAD1600吸附树脂(φ8*80cm),装量为1500ml,上样流速为2700ml/h。树脂用12L纯化水水洗,流速1800ml/h,然后用12L,5%的乙醇水溶液解吸,流速2700ml/h。每隔300ml收集一个解吸组分,并将纯度在90%以上的组分混合,得到7.2L解吸混合液。向解吸混合液中加入767针用活性炭7.2g,搅拌30min后过滤得到脱色液。将脱色液进行纳滤浓缩,浓缩至380ml。再将纳滤液用旋转蒸发器在45℃下浓缩至13ml,然后浓缩液在4℃下搅拌结晶2h后,得到13.52g潮晶(HPLC的图谱见图2和表2),纯度为98.4%。然后用8ml纯化水将潮晶溶解,于9℃下搅拌过夜,抽滤,得到的白色晶体进行真空加热干燥,得到9.60g二次结晶FR179642精粉(HPLC的图谱见图3和表3),纯度为99.8%,总收率为60.4%。表1:FR179642滤液的HPLC谱图数据峰保留时间面积峰高%面积11.1874433946780.12022.195451479927720712.26032.52114601721248083.96542.907797674539462.16653.312646738583401.75663.475520249512621.41374.11818379091252754.99185.85925174136136841468.36197.0501872110090.051107.3831768210970.048118.2015888524080.160129.2653308014440.0901310.46810744738730.2921413.5211441593319043.9151515.6863519412260.0961618.97411649826410.316总计368251152109533100.000表2:FR179642一次潮晶的HPLC谱图数据峰保留时间面积峰高%面积13.38326242810.01923.823181123173461.28334.15045584930.03245.2821389919394267098.44856.167129600.00966.96450043000.035711.121245098100.174总计14118308961901100.00表3:FR179642精粉的HPLC谱图数据峰保留时间面积峰高%面积13.8611112213590.17724.111441490.00735.349627269746745299.80747.068582430.009总计6284842468903100.00实施例2发酵液FR179642放罐73.6L,发酵单位为628μg/ml。向其中加入1.5kg硅藻土,搅拌半小时后板框压滤,得到62.1L滤液(HPLC峰型与图1相似)。将滤液进行旋转蒸发浓缩,得到浓缩液13.3L,发酵单位为3642μg/ml。将浓缩液导入XAD1600吸附树脂(φ15*100cm),装量为3.4L,上样流速为10.8L/h。树脂用80L纯化水水洗,流速13.2L/h,然后用26L,5%的丙酮水溶液解吸,流速13.2ml/h。每隔600ml收集一个解吸组分,并将纯度在90%以上的组分混合,得到解吸混合液22.4L。向解吸混合液中加入767针用活性炭20g,搅拌脱色处理30min后过滤得到脱色液。然后将脱色液进行纳滤浓缩,浓缩至5600ml,再将纳滤液用旋转蒸发器在45℃下浓缩至600ml。然后浓缩液在4℃下搅拌结晶2h后,得到28.6g潮晶(HPLC的峰型与图2相似),纯度为98.7%。然后用50ml纯化水将潮晶50℃下搅拌20min,再在室温条件下搅拌30min,最后于8℃下搅拌过夜,抽滤得到的白色晶体进行真空加热干燥,得到26.7g二次结晶FR179642精粉(HPLC的峰型与图3相似),纯度为99.3%,总收率为58.3%。实施例3发酵液FR179642放罐4.9L,发酵单位为1350μg/ml。向其中加入0.2kg珍珠岩,搅拌半小时后,抽滤分离,得到4.7L滤液(HPLC峰型与图1相似)。将滤液进行闪蒸浓缩,得到浓缩液1.5L,发酵单位为4410μg/ml。将浓缩液导入XAD1180吸附树脂(φ4*50cm),装量为790ml,上样流速为1000ml/h。树脂用4L纯化水水洗,流速1000ml/h,然后用15L,15%的乙醇水溶液解吸,流速800ml/h。每隔200ml收集一个解吸组分,并将纯度在90%以上的组分混合,得到解吸混合液1.4L。向解吸混合液中加入767针用活性炭1.5g,搅拌脱色处理25min后,过滤,得到脱色液。然后将脱色液进行纳滤浓缩至350ml后用旋转蒸发器在45℃下浓缩至10ml,然后浓缩液在8℃下搅拌结晶3h后,得到4.7g潮晶(HPLC峰型与图2相似),纯度为98.5%。再用4ml纯化水将潮晶溶解,于10℃下搅拌过夜,抽滤,得到的晶体进行真空加热干燥,得到4.12g二次结晶FR179642精粉(HPLC峰型与图3相似),纯度为99.1%,总收率为62.3%。实施例4发酵液FR179642放罐34.3L,发酵单位为850μg/ml。向其中加入2.1kg硅藻土,搅拌半小时后抽滤分离,得到28.5L滤液(HPLC峰型与图1相似)。将滤液进行闪蒸浓缩,得到浓缩液5000ml,发酵单位为,5830μg/ml。将浓缩液导入XAD1180吸附树脂(φ8*80cm),装量为2.0L,上样流速为2400ml/h。树脂用12L纯化水水洗,流速2000ml/h,然后用12L,15%的丙酮水溶液解吸,流速2400ml/h。每隔500ml收集一个解吸组分,并将纯度在90%以上的组分混合,得到解吸混合液10.9L。向解吸混合液中加入767针用活性炭10.5g,搅拌脱色处理25min后,过滤,得到脱色液。然后将脱色液进行纳滤浓缩,浓缩至650ml。再将纳滤液用旋转蒸发器在45℃下浓缩至16ml后在8℃下搅拌结晶4h后,得到21.5g潮晶(HPLC峰型与图2相似),纯度为98.3%。然后用12ml纯化水将潮晶溶解,于4℃下搅拌过夜,抽滤,得到的白色晶体进行真空加热干燥,得到17.3gFR179642精粉(HPLC峰型与图3相似),纯度为99.0%,总收率为59.3%。对比实施例1含有FR179642发酵液22.7L,发酵单位为700μg/ml。向其中加入1kg硅藻土,搅拌半小时后离心分离,得到18.9L滤液。将滤液进行闪蒸浓缩,得到浓缩液3.8L,单位为3642μg/ml。将浓缩液导入XAD1600吸附树脂(φ8*80cm),装量为1500ml,上样流速为2700ml/h。树脂用20%的乙醇洗涤,流速1800ml/h,然后用50%的乙醇水溶液解吸,流速2700ml/h。每隔300ml收集一个解吸组分,并将纯度在90%以上的组分混合,组成解吸混合液。然后将解吸混合液进行纳滤浓缩,浓缩至380ml。最后将纳滤液用旋转蒸发器在45℃下浓缩至13ml,纯度为90.2%,总收率为31.8%。对比实施例2含有FR179642发酵液22.7L,发酵单位为700μg/ml。向其中加入1kg硅藻土,搅拌半小时后离心分离,得到18.9L滤液。将滤液进行闪蒸浓缩,得到浓缩液3.8L,单位为3642μg/ml。将浓缩液导入XAD1600吸附树脂(φ8*80cm),装量为1500ml,上样流速为2700ml/h。树脂用40%的乙醇洗涤,流速1800ml/h,然后用60%的乙醇水溶液解吸,流速2700ml/h。每隔300ml收集一个解吸组分,并将纯度在90%以上的组分混合,组成解吸混合液。然后将解吸混合液进行纳滤浓缩,浓缩至380ml。最后将纳滤液用旋转蒸发器在45℃下浓缩至13ml,纯度为90.5%,总收率为30.2%。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1