一种右旋兰索拉唑晶型化合物及其制备方法与流程
本发明属于医药
技术领域:
,具体地说,涉及一种右旋兰索拉唑晶体化合物及其制备方法。
背景技术:
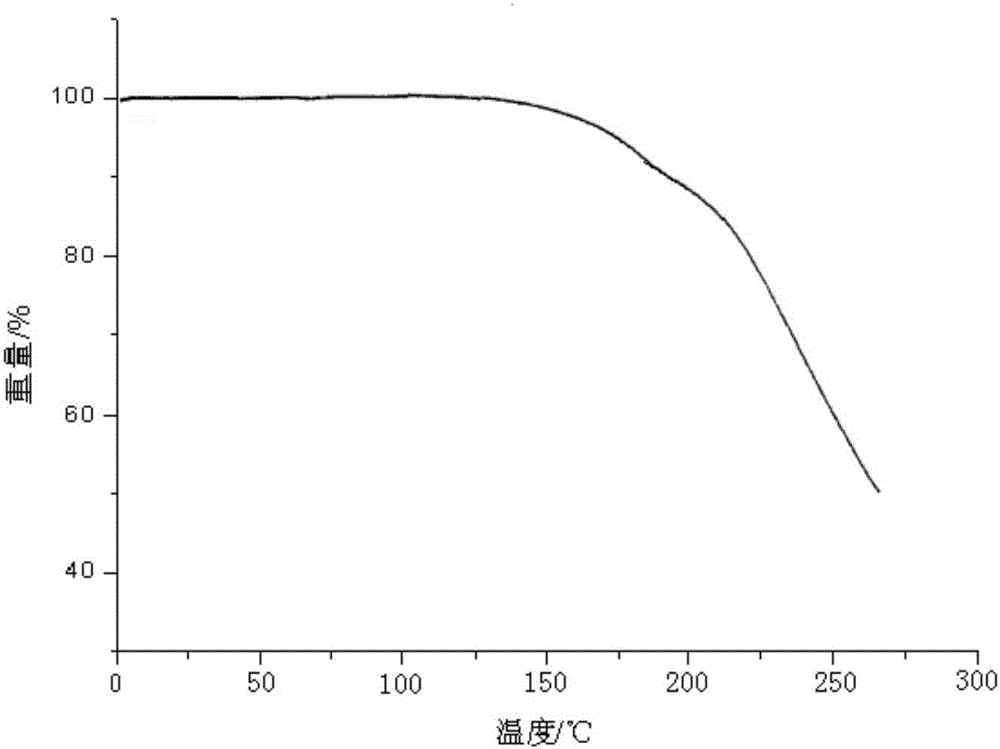
:兰索拉唑(lansoprazole)是由武田公司开发的世界上第二个质子泵抑制剂抗溃疡药。1992年投放市场,2004年6月在美国获准上市。兰索拉唑通过抑制胃粘膜壁细胞质子泵,及抑制H+、K+-ATP酶的活性,使壁细胞的H+不能转运到胃中去,从而持续地抑制胃酸和胃蛋白酶的分泌。兰索拉唑因在吡啶环4位侧链导入氟,而且有三氟乙氧基取代基,使其生物利用度较奥美拉唑提高30%以上,亲脂性也强于奥美拉唑,因此该品在酸性条件下可迅速地透过壁细胞膜转变为次磺酸和次磺酸衍生物而发挥药效,对HP的抑菌活性提高至奥美拉唑的4倍。右旋兰索拉唑是质子泵抑制剂兰索拉唑的对映体,其活性明显高于消旋的兰索拉唑,毒副作用低于消旋的兰索拉唑。目前已经在国外上市。现有的结果表明:右旋兰索拉唑的药效好于左旋构型的兰索拉唑,消旋体的药效主要来自右旋兰索拉唑。右旋兰索拉唑钠的化学结构如下:右兰索拉唑有多种晶型,现有技术中也公开了右旋兰索拉唑的多种晶型,比如WO2000/78745(同族专利CN1150186C)公开了一种无水晶型和一种含1.5个结晶水的水合物晶型,CN102234265A公开了右旋兰索拉唑的两个半结晶水晶型,CN102875531A公开了一种(R)-兰索拉唑无水晶型及其制备方法,CN104086532A公开了右兰索拉唑晶型α、晶型β和晶型γ。专利CN102875531B研究的发现,(R)-兰索拉唑对水分敏感,在空气中放置极易吸收水分。现有技术中晶型水合物稳定性差,容易降解,寻求制备右旋兰索拉唑的无水晶型是非常有必要的。专利CN1150186C(无水晶型称为A)和CN102875531B(无水晶型称为B),令人惊奇地,发现本发明制备制得第三种无水晶型,称作晶型C,它在热力学上比晶型A更加稳定,且具有比已经晶型B更好的溶解度。所以本发明提供新的右旋兰索拉唑晶型,它意外的高于晶型B预期溶解度,并且与多晶型A相比提高了热力学稳定性。本发明的晶型与已知晶型A和B相比具有改进的药理学性质。技术实现要素:本发明的目的提供了一种右旋兰索拉唑晶型化合物,该晶型稳定性高,水溶性得到有效改善。本发明的另一个目的还在于公开了右旋兰索拉唑晶型化合物的制备方法。为实现本发明的目的,本发明采用如下技术方案:一种式(I)所示的右旋兰索拉唑晶型化合物,其中,所述的右旋兰索拉唑晶型化合物使用Cu-Kα射线测量得到的X-射线粉末衍射谱图如图1所示。本发明还进一步提供所述的右旋兰索拉唑晶型化合物的制备方法,该方法包括如下步骤:1)将右旋兰索拉唑粗品加入醇类溶剂与卤代烃类混合溶剂于室温进行搅拌溶解;2)待步骤1)中溶清后过滤,冷却至-10℃~10℃,养晶2~12小时,制得右旋兰索拉唑晶型。作为优选的,步骤1)中所述醇类溶剂为甲醇、乙醇或异丙醇。作为优选的,步骤2)中所述卤代烃类溶剂为二氯甲烷或三氯甲烷。作为优选的,所述醇类溶剂与卤代烃类溶剂的体积比为1∶1~6。本发明人经过大量的反复试验,不断改变结晶方法以及包括溶剂、温度等结晶条件,最终得到一种右旋兰索拉唑的新晶型,该新晶型的右旋兰索拉唑具有较好的稳定性,且相比于现有技术的右旋兰索拉唑晶型改善了水中溶解度。附图说明图1为本发明实施例1所制备的右旋兰索拉唑晶型化合物的X射线粉末衍射图;图2为本发明实施例1所制备的右旋兰索拉唑晶型化合物的热重分析图谱。具体实施方式通过下面的实施例可以对本发明进行进一步的描述,然而,本发明的发明并不限于下面的实施例,这些实施例不以任何方式限制本发明的范围。本领域的技术人员在权利要求的范围内所作出的某些改变和调整也应认为属于本发明的范围。实施例1右旋兰索拉唑晶型化合物的制备在洁净干燥的100ml四口烧瓶中,投入右旋兰索拉唑粗品10g、甲醇10ml、二氯甲烷10ml,室温搅拌溶清后过滤,滤液冷却至-10~-5℃,有少量晶体析出,养晶2h,过滤,湿品放入45℃真空干燥得到终产品右旋兰索拉唑晶型化合物。所制得的右旋兰索拉唑用粉末X射线衍射测定法测定,得到X-射线粉末衍射图谱如图1所示。采用美国Perkin-Elmer公司PEPyrisDiamondTG热重分析仪,热重分析图谱如图2所示,晶型熔点为145-151℃,右旋兰索拉唑晶型在降解之前无重量损失,采用卡式水分测定水含量为0.12wt%,可以推断为右旋兰索拉唑的无水合物。实施例2右旋兰索拉唑晶型化合物的制备在洁净干燥的100ml四口烧瓶中,投入右旋兰索拉唑粗品10g、甲醇10ml、三氯甲烷30ml,室温搅拌溶清后过滤,滤液冷却至-10~-5℃,有少量晶体析出,养晶12h,过滤,湿品放入45℃真空干燥得到终产品右旋兰索拉唑晶型化合物。根据XPRD数据,所得晶型与实施例1中晶型一致。采用美国Perkin-Elmer公司PEPyrisDiamondTG热重分析仪得到的热重分析图谱与实施例1一致。实施例3右旋兰索拉唑晶型化合物的制备在洁净干燥的100ml四口烧瓶中,投入右旋兰索拉唑粗品10g、乙醇10ml、二氯甲烷60ml,室温搅拌溶清后过滤,滤液冷却至-5~0℃,有少量晶体析出,养晶7h,过滤,湿品放入45℃真空干燥得到终产品右旋兰索拉唑晶型化合物。根据XPRD数据,所得晶型与实施例1中晶型一致。采用美国Perkin-Elmer公司PEPyrisDiamondTG热重分析仪得到的热重分析图谱与实施例1一致。实施例4右旋兰索拉唑晶型化合物的制备在洁净干燥的100ml四口烧瓶中,投入右旋兰索拉唑粗品10g、异丙醇10ml、二氯甲烷60ml,室温搅拌溶清后过滤,滤液冷却至-10~-5℃,有少量晶体析出,养晶2h,过滤,湿品放入45℃真空干燥得到终产品右旋兰索拉唑晶型化合物。根据XPRD数据,所得晶型与实施例1中晶型一致。采用美国Perkin-Elmer公司PEPyrisDiamondTG热重分析仪得到的热重分析图谱与实施例1一致。试验例11、右旋兰索拉唑晶型晶型在高温、高湿、强光条件下的稳定性加速试验。1.1高温影响因素实验:分别取1g右旋兰索拉唑A、B、C晶型,置于洁净容器中,在温度60℃,相对湿度75%的条件下放置10天,分别在0、5、10天取样,观察其外观、色泽测定杂质含量,结果详见表1。表1:右旋兰索拉唑晶型高温影响因素实验结果晶型A:按照专利CN1150186C实施例2制备的右旋兰索拉唑晶型。晶型B:按照专利CN102875531B实施例1制备的右旋兰索拉唑晶型。晶型C:按照本发明实施例1制备的右旋兰索拉唑晶型。结果表明,右旋兰索拉唑在高温条件下放置10天后,从性状、外观上看右旋兰索拉唑晶型A、B颜色变深,右旋兰索拉唑晶型C基本没变化,由表1可见,右旋兰索拉唑C晶型与其它右旋兰索拉唑晶型相比,右旋兰索拉唑C晶型在高温条件下放置10天后其杂质的含量明显低于其它右旋兰索拉唑晶型。1.2高湿影响因素实验分别取1g右旋兰索拉唑A、B、C晶型,在含饱和硝酸钾溶液的干燥器中(25℃,相对湿度92.5%)放置10天,分别在第0、5、10天取样,测定杂质含量,结果见表2。表2:右旋兰索拉唑晶型高湿影响因素实验结果结果表明,由表2可见,右旋兰索拉唑C晶型与其它右旋兰索拉唑晶型相比,右旋兰索拉唑X晶型在高湿条件下放置10天后其杂质的含量明显低于其他右旋兰索拉唑晶形。1.3强光影响因素实验:分别取1g右旋兰索拉唑A、B、C晶型,在照度为(4500±500)1x的日光灯的光照箱内放置10天,分别在第0、5、10天取样,结果见表3。表3:右旋兰索拉唑晶型强光影响因素试验结果结果表明,由表3可知,右旋兰索拉唑C晶型与其它右旋兰索拉唑晶型相比,右旋兰索拉唑C晶型在强光条件下放置10天后其杂质的含量明显低于其它右旋兰索拉唑晶型。2、右旋兰索拉唑晶型稳定性试验分别取1g右旋兰索拉唑A、B、C晶型,置于洁净容器中,在温度40℃,相对湿度75%的条件下放置6个月,第6个月取样,考察右旋兰索拉唑晶型杂质及含量等指标,实验结果见表4。表4:右旋兰索拉唑加速6个月注:澄清度与颜色“澄明无色为合格、可见异物≤4个为合格,≥25um的不溶性微粒≤600个为合格)。结果表明:由表4可见,右旋兰索拉唑A、B、C晶型6个月加速实验后,右旋兰索拉唑C晶型与其他右旋兰索拉唑晶型相比,右旋兰索拉唑C晶型性状基本没变化,澄明度与颜色、可见异物、不溶性微粒均合格,其他右旋兰索拉唑晶型均不合格;右旋兰索拉唑C晶型有关物质均明显小于其他右旋兰索拉唑晶型,含量明显高于其他右旋兰索拉唑晶型。2、溶解度试验该试验例考察了本发明的右旋兰索拉唑晶型化合物与现有技术的右旋兰索拉唑在水中的溶解性。溶解度试验方法:按照中国药典2010年版二部“凡例”规定的方法做了以下几种溶剂的溶解度试验。方法:称取研成细粉的各样品适量(精确至±2.0%),加入一定量体积的水,在25±2℃室温下每隔5分钟振摇30秒,观察30分钟内溶解情况,结果见表1。表5:溶解度试验结果晶型种类25℃溶解度(mg/ml)右旋兰索拉唑晶型A1.91右旋兰索拉唑晶型B2.57右旋兰索拉唑晶型C28.5从试验结果可以看出,本发明与现有技术相比,本发明制得的右旋兰索拉唑晶型化合物在水中的溶解性高于现有技术右旋兰索拉唑无水晶型。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1