运用CRISPR‑Cas9系统敲除大麦VE合成通路中的关键基因HPT的方法与流程
本发明涉及大麦转基因材料的构建,特别涉及运用CRISPR-Cas9系统敲除HvHPT(MLOC_37476)基因的应用。
背景技术:
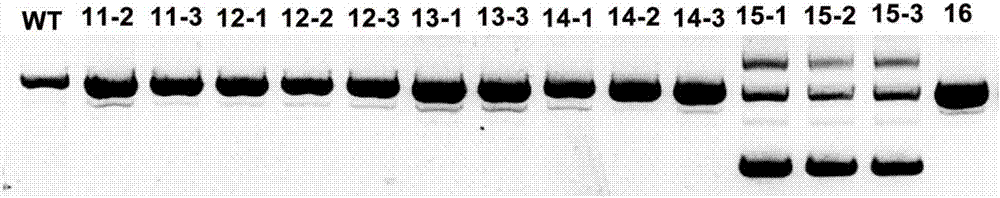
:维生素E(vitaminE,VE)是由光合生物合成的生育酚类化合物的总称。根据侧链是否饱和,VE可以分为生育酚(tocophero1)和生育三烯酚(tocotrieno1)两大类。根据芳香环上甲基位置和数目的不同,每类又可分为α,β,γ,δ四种形式,其中,α-生育酚活性最高。近些年来,由于生育三烯酚在某些方面更优越的生物学特性,备受人们关注。不仅表现在抗氧化活性,在降胆固醇、预防糖尿病、促进骨吸收、抗癌、神经保护等方面也有一定的作用。大麦是世界上四大粮食作物之一,主要用于食品生产、动物饲养、啤酒制造等领域。另外,由于大麦含有丰富的生物活性物质如β-葡聚糖、酚类物质、维生素E等,也常被用作功能食品开发的原料。大麦谷粒,含有丰富的生育三烯酚,大概占VE总含量的70%,是研究生育三烯酚的好材料。因此利用基因工程手段调控大麦谷粒中的VE合成通路,可以提高大麦生育三烯酚的含量,从而起到增加大麦谷粒营养成分的作用。目前由于大麦突变体库的缺乏,限制了大麦VE合成通路中相关基因的功能研究。近年来发展起来的以CRISPR-Cas9为代表的新一代基因组编辑技术,为植物基因工程带来了新的革命,已成为基因功能研究和作物品质改良的重要手段之一。运用基于CRISPR-Cas9的基因编辑技术改造相关基因,并通过自交、分子鉴定和后代筛选,获得“非转基因”大麦新材料,可为今后将成果应用于生产实践提供依据和技术支持。但是因为大麦基因转化效率低,稳定转基因材料的获得周期长,目前运用CRISPR-Cas9系统研究大麦基因的报导很少见。因此,应用CRISPR-Cas9技术敲除大麦VE合成通路中的关键基因(HvHPT)而获得的大麦突变体,可为HvHPT基因的功能研究提供可靠材料。技术实现要素:本发明要解决的问题是,克服现有技术中的不足,提供一种运用CRISPR-Cas9系统敲除大麦HPT基因的编辑方法,以获得HPT基因突变的理想突变体。为解决技术问题,本发明的解决方案是:提供一种运用CRISPR-Cas9系统敲除大麦HPT基因的编辑方法,包括以下步骤:(1)gRNA靶位点的选择由于HPT基因位于大麦基因组的七号染色体上,根据CRISPR-Cas9技术的靶位点设计原则,应尽量将gRNA靶位点设计在外显子区并且要设计在基因的5’端(因该基因编码的是蛋白质,5’端编码的正好是蛋白质的功能区域)。(2)gRNA片段的克隆以质粒pGTR为模板,用PCR方法克隆四个片段L1、L2、L3、L4部分重叠的片段,引物序列如下,其中F和R分别代表正、反向引物:L1-F:CGGGTCTCAGGCAGGATGGGCAGTCTGGGCAACAAAGCACCAGTGGL1-R:CGGGTCTCACCCCTACCCTATTGCACCAGCCGGGL2-F:TAGGTCTCCGGGGGTAGGGGTGTTTTAGAGCTAGAAL2-R:CGGGTCTCACATACTGTTCCTTGCACCAGCCGGGL3-F:TAGGTCTCCTATGCCGAAACGGTTTTAGAGCTAGAAL3-R:CGGGTCTCACAGTATCGTGTGTGCACCAGCCGGGL4-F:TAGGTCTCCACTGCAAGCTTCGTTTTAGAGCTAGAAL4-R:TAGGTCTCCAAACGGATGAGCGACAGCAAACAAAAAAAAAAGCACCGACTCGPCR体系为:Phusion酶0.5μL;5×PhusionHFBuffer10μL;上下游引物各2.5μL;dNTPs4μL;pGTRplasmid0.5μL;三蒸水30μL,共50μL体系。PCR反应条件为:预变性95℃,5min;变性95℃,30s;退火60℃,30s;延伸72℃,30s;共33个循环;最后72℃延伸10min;PCR反应后,取5-10μL产物,用2%的琼脂糖凝胶电泳检测后,纯化回收目的片段,测定四个PCR产物L1、L2、L3、L4的浓度;(3)GG(gRNA-gRNA)片段的连接根据测定的PCR产物浓度,将四个片段等量混合,T7酶连接反应与BsaI酶切反应同时进行;取L1、L2、L3、L4各2μL,与10μLT7ligasebuffer、1μLBsaI-HF、0.5μLT7ligase、0.5μL水混合;在PCR仪中进行如下反应:37℃,5min;20℃,10min;30-50个循环;(4)连接产物的PCR扩增;连接反应结束后,取连接产物1μL,加19μL水稀释,将稀释后的产物作为模板,进行PCR扩增;PCR结束后,取5μL产物进行电泳检测,并将产物纯化;产物大小为500bp;引物序列如下,其中F和R分别代表正、反向引物:S1-F:CGGGTCTCAGGCAGGATGGGCAGTCTGGGCAS1-R:TAGGTCTCCAAACGGATGAGCGACAGCAAAC(5)纯化产物和目标载体的酶切FokI酶切纯化产物暴露黏性末端,同时BsaI酶切空载体pRGEB32;酶切体系为50μL,包括底物5μL、FokI或BsaI酶5μL、Buffer(cutsmart)10μL;底物包括GG纯化产物和空载体pRGEB32;酶切时间3-4h,酶切温度37℃;用2%琼脂糖凝胶检测酶切产物,并回收目标产物,测定浓度;(6)纯化产物和目标载体的连接反应取酶切回收的GG纯化产物与pRGEB32载体等量混合(50ng),T4DNAligase1μL,10×T4DNAligaseBuffer1μL,加三蒸水至20μL,4℃连接过夜;(7)连接产物转化大肠杆菌感受态将连接后的载体转化大肠杆菌感受态细胞,涂板,37℃过夜;挑取单菌落,摇菌6-8h,提取质粒,PCR鉴定目标片段是否连入载体;鉴定正确的质粒送去测序,将测序结果正确的质粒,电转化农杆菌AGL1;(8)农杆菌介导的大麦转基因以GoldenPromise野生型大麦的幼胚为外植体材料,以AGL1农杆菌进行感染转化,经潮霉素抗性筛选,抗性愈伤组织分化再生获得转基因植株;(9)阳性转基因植株的筛选提取转基因植株的基因组DNA,在三个gRNA序列的两侧设计引物,对目的片段进行PCR扩增,利用琼脂糖凝胶电泳和垂直聚丙烯凝胶电泳检测突变体;(10)突变体测序将以上突变株系PCR产物进行纯化回收,连接T载体测序,确认获得敲除了大麦HPT基因的突变材料。发明原理描述:VE的合成通路比较复杂,生育酚和生育三烯酚有共同的合成前体尿黑酸(homogentisate,HGA)。其中生育三烯酚合成的限速步骤是由尿黑酸牻牛儿基转移酶(homogentisategeranylgeranyltransferase,HGGT)催化HGA和牻牛儿焦磷酸(geranylgeranyldiphosphate,GGDP)的合成反应;而生育酚合成的限速步骤是由尿黑酸植基转移酶(homogentisatephytyltransferase,HPT)催化HGA和植基二磷酸(phytyldiphosphate,PDP)的合成反应。HGGT、HPT分别是生育三烯酚、生育酚合成过程中的关键基因,因此可以通过敲除生育酚合成通路中的关键限速酶基因HPT来调节代谢流,从而起到提高或者分离生育三烯酚组分的作用。CRISPR-Cas系统可定点修饰(删除、添加、激活、抑制)靶细胞中特定的基因序列,为靶向性编辑基因组序列提供行之有效的技术手段。但是,目前尚无运用该技术敲除大麦VE合成通路中的关键基因HPT的报道。与现有技术相比,本发明的有益效果在于:本发明利用先进的基因编辑技术-CRISPR-Cas9系统对大麦VE合成通路中的关键基因(HPT)进行靶向基因编辑,获得有效的功能缺失突变体,为大麦中生物活性物质的研究创造条件。附图说明图1为靶向大麦HPT基因的3个gRNA位点示意图;图2为琼脂糖凝胶电泳检测大片段缺失突变体;图3为PAGE凝胶电泳检测突变体;图4为突变株系目的片段测序结果的比较(点状虚线表示缺失碱基,单下划线表示插入碱基)。具体实施方式实施例1大麦VE合成通路中的关键限速酶基因敲除株系HPT的获得与鉴定。本发明所用大麦品种为HordeumvulgareL.,cv.Goldenpromise,申请人承诺:从本专利申请之日起20年内向公众发放该大麦品种,以用于实现、利用本发明所述技术方案。1.gRNA靶位点的选择由于HPT基因位于大麦基因组的七号染色体上,根据CRISPR-Cas9技术的靶位点设计原则,本发明将gRNA靶位点设计在外显子区并且要设计在HPT基因的5’端(因该基因编码的是蛋白质,5’端编码的正好是蛋白质的功能区域)。如图1所示。2.gRNA片段的克隆及载体构建2.1以质粒pGTR为模板,用PCR方法克隆四个片段L1、L2、L3、L4部分重叠的片段,引物序列如下:L1-F:CGGGTCTCAGGCAGGATGGGCAGTCTGGGCAACAAAGCACCAGTGG(如SEQIDNO:1所示)L1-R:CGGGTCTCACCCCTACCCTATTGCACCAGCCGGG(如SEQIDNO:2所示)L2-F:TAGGTCTCCGGGGGTAGGGGTGTTTTAGAGCTAGAA(如SEQIDNO:3所示)L2-R:CGGGTCTCACATACTGTTCCTTGCACCAGCCGGG(如SEQIDNO:4所示)L3-F:TAGGTCTCCTATGCCGAAACGGTTTTAGAGCTAGAA(如SEQIDNO:5所示)L3-R:CGGGTCTCACAGTATCGTGTGTGCACCAGCCGGG(如SEQIDNO:6所示)L4-F:TAGGTCTCCACTGCAAGCTTCGTTTTAGAGCTAGAA(如SEQIDNO:7所示)L4-R:TAGGTCTCCAAACGGATGAGCGACAGCAAACAAAAAAAAAAGCACCGACTCG(如SEQIDNO:8所示)PCR扩增L1片段体系如下:Phusion酶0.5μL5×Buffer10μLL1-F2.5μLL1-R2.5μL三蒸水30μLdNTPs4μL模板(pGTRplasmid)0.5μL总计50μLPCR扩增L2片段体系如下:Phusion酶0.5μL5×Buffer10μLL2-F2.5μLL2-R2.5μL三蒸水30μLdNTPs4μL模板(pGTRplasmid)0.5μL总计50μLPCR扩增L3片段体系如下:PCR扩增L4片段体系如下:Phusion酶0.5μL5×Buffer10μLL4-F2.5μLL4-R2.5μL三蒸水30μLdNTPs4μL模板(pGTRplasmid)0.5μL总计50μLPCR反应程序为:PCR反应后,取5-10μL产物,用2%的琼脂糖凝胶电泳检测后纯化回收目的片段,测定产物浓度。L1,L2,L3,L4产物大小约为130bp,200bp,200bp,150bp。2.2GG(gRNA-gRNA)片段的连接。按照上步测定的产物浓度,将4个片段等量混合,T7酶连接。反应体系如下:试剂体积(μL)L12L22L32L422×T7ligasebuffer10BsaI-HF1T7ligase0.5三蒸水0.5总体积20以上连接反应在PCR仪中进行:37℃,5min;20℃,10min;30-50个循环。2.3连接产物的PCR扩增连接反应结束后,取连接产物1μL,加19μL水稀释,将稀释后的产物作为模板,以S1-F、S1-R为引物进行PCR扩增。S1-F:CGGGTCTCAGGCAGGATGGGCAGTCTGGGCA(如SEQIDNO:9所示)S1-R:TAGGTCTCCAAACGGATGAGCGACAGCAAAC(如SEQIDNO:10所示)PCR体系如下:试剂体积(μL)稀释后的GG产物2.5S1-F2.5S1-R2.52×TaqMix25三蒸水17.5总体积50PCR反应程序为:取5-10μLPCR产物电泳检测,产物大小约为500bp,并将产物纯化。2.4将上一步纯化的产物,FokI酶切暴露黏性末端,同时BsaI酶切空载体pRGEB32。酶切体系为50μL,包括底物5μL、FokI或BsaI酶5μL、Buffer(cutsmart)10μL;底物包括GG纯化产物和空载体pRGEB32;酶切时间3-4h,作用温度37℃;用2%琼脂糖凝胶检测酶切产物,并回收目标产物,测定浓度;2.5酶切后的GG连接产物与目标载体pRGEB32的连接。试剂体积(μL)酶切后的GG连接产物50ng酶切后的载体pRGEB3250ngT4DNAligase110×T4ligasebuffer1三蒸水补至10总体积103.将连接后的载体转化大肠杆菌感受态细胞,涂板,37℃过夜;挑取单菌落,摇菌6-8h,提取质粒,PCR鉴定目标片段是否连入载体;鉴定正确的质粒送去测序。PCR鉴定及测序引物为U3-F,UGW-gRNA-R。序列如下:U3-F:AGTACCACCTCGGCTATCCACA(如SEQIDNO:11所示)UGW-gRNA-R:CGCGCTAAAAACGGACTAGC(如SEQIDNO:12所示)4.将测序正确的质粒,电转化农杆菌AGL1。5.农杆菌介导的大麦的遗传转化以野生型大麦(HordeumvulgareL.,cv.GoldenPromise)的幼胚为材料诱导愈伤组织,以AGL1农杆菌进行感染转化,经潮霉素抗性筛选,抗性愈伤组织分化再生获得转基因植株;6.阳性转基因植株的筛选6.1琼脂糖凝胶电泳检测大片段缺失突变体以叶片为材料,提取转基因植株的基因组DNA,在3个gRNA位点的两侧设计引物进行PCR扩增,1%琼脂糖凝胶电泳检测。(结果见图2)引物序列如下所示:H1-F:ACCTTTCAGTCAGTGGCTTTGAACT(如SEQIDNO:13所示)H2-R:ACCTCCAGCAATCCAGTAAG(如SEQIDNO:14所示)6.2PAGE胶检测小片段变化的突变体以叶片为材料,提取转基因植株的基因组DNA,在每个gRNA位点两侧设计引物进行PCR。在常规PCR反应结束后,再对PCR产物进行高温变性、复性反应,PCR程序和变性复性步骤如下表:引物如下所示:H1-F:ACCTTTCAGTCAGTGGCTTTGAACT(如SEQIDNO:13所示)H1-R:TTACAAGAGGCGTTGCTGGTTCATT(如SEQIDNO:15所示)H2-F:CCACAACAAATCTACCGTCTC(如SEQIDNO:16所示)H2-R:ACCTCCAGCAATCCAGTAAG(如SEQIDNO:14所示)结果见图3。7.突变株系的基因测序将以上突变株系PCR产物进行纯化回收。大片段缺失的突变体可直接割胶回收测序,小片段变化的突变体则需连接T载体进行测序。测序公司为杭州擎科梓熙生物技术有限公司。测序结果见图4。测序结果分析,发现了15#株系存在746bp的大片段缺失,获得了敲除大麦HPT基因的理想突变材料。<110>浙江大学<120>运用CRISPR-Cas9系统敲除大麦VE合成通路中的关键基因HPT的方法<160>16SEQIDNO:1<210>1<211>46<212>DNA<213>人工序列<220><223>用于PCR克隆片段L1的正向引物L1-F<400>1CGGGTCTCAGGCAGGATGGGCAGTCTGGGCAACAAAGCACCAGTGG46SEQIDNO:2<210>2<211>34<212>DNA<213>人工序列<220><223>用于PCR克隆片段L1的反向引物L1-R<400>2CGGGTCTCACCCCTACCCTATTGCACCAGCCGGG34SEQIDNO:3<210>3<211>36<212>DNA<213>人工序列<220><223>用于PCR克隆片段L2的正向引物L2-F<400>3TAGGTCTCCGGGGGTAGGGGTGTTTTAGAGCTAGAA36SEQIDNO:4<210>4<211>34<212>DNA<213>人工序列<220><223>用于PCR克隆片段L2的反向引物L2-R<400>4CGGGTCTCACATACTGTTCCTTGCACCAGCCGGG34SEQIDNO:5<210>5<211>36<212>DNA<213>人工序列<220><223>用于PCR克隆片段L3的正向引物L3-F<400>5TAGGTCTCCTATGCCGAAACGGTTTTAGAGCTAGAA36SEQIDNO:6<210>6<211>34<212>DNA<213>人工序列<220><223>用于PCR克隆片段L3的反向引物L3-R<400>6CGGGTCTCACAGTATCGTGTGTGCACCAGCCGGG34SEQIDNO:7<210>7<211>36<212>DNA<213>人工序列<220><223>用于PCR克隆片段L4的正向引物L4-F<400>7TAGGTCTCCACTGCAAGCTTCGTTTTAGAGCTAGAA36SEQIDNO:8<210>8<211>52<212>DNA<213>人工序列<220><223>用于PCR克隆片段L4的反向引物L4-R<400>8TAGGTCTCCAAACGGATGAGCGACAGCAAACAAAAAAAAAAGCACCGACTCG52SEQIDNO:9<210>9<211>31<212>DNA<213>人工序列<220><223>用于连接产物PCR扩增正向引物S1-F<400>9CGGGTCTCAGGCAGGATGGGCAGTCTGGGCA31SEQIDNO:10<210>10<211>31<212>DNA<213>人工序列<220><223>用于连接产物PCR扩增反向引物S1-R<400>10TAGGTCTCCAAACGGATGAGCGACAGCAAAC31SEQIDNO:11<210>11<211>22<212>DNA<213>人工序列<220><223>用于PCR鉴定及测序的引物U3-F<400>11AGTACCACCTCGGCTATCCACA22SEQIDNO:12<210>12<211>20<212>DNA<213>人工序列<220><223>用于PCR鉴定及测序的引物UGW-gRNA-R<400>12CGCGCTAAAAACGGACTAGC20SEQIDNO:13<210>13<211>25<212>DNA<213>人工序列<220><223>用于电泳检测的PCR扩增引物H1-F<400>13ACCTTTCAGTCAGTGGCTTTGAACT25SEQIDNO:14<210>14<211>20<212>DNA<213>人工序列<220><223>用于电泳检测的PCR扩增引物H2-R<400>14ACCTCCAGCAATCCAGTAAG20SEQIDNO:15<210>15<211>25<212>DNA<213>人工序列<220><223>用于PAGE胶检测的PCR扩增引物H1-R<400>15TTACAAGAGGCGTTGCTGGTTCATT25SEQIDNO:16<210>16<211>21<212>DNA<213>人工序列<220><223>用于PAGE胶检测的PCR扩增引物H2-F<400>16CCACAACAAATCTACCGTCTC21当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1