孢子丝菌DRK1基因GFP报告菌株的构建方法与流程
本发明属于真菌报告菌株构建领域。具体涉及孢子丝菌DRK1基因启动子的获得、DRK1基因终止子的获得、重组载体PCB309-PGFPT的构建、用于转染的根癌农杆菌及孢子丝菌。
背景技术:
孢子丝菌病是由申克孢子丝菌及其复合体引起的皮肤、皮下组织及附近淋巴管的慢性感染性疾病,是最常见的皮肤深部真菌病。随着宠物饲养及免疫受损人群数量的增多,本病的发病率不断攀升,并出现局部区域的爆发流行。孢子丝菌病在全球范围内广泛分布,我国为该病的高发地区,2008-2013年间仅一家医院就接诊孢子丝菌病患者1500余例,严重危害民众健康。
孢子丝菌属于双相病原真菌,此类真菌在自然环境中呈腐生状态生长,无致病性,入侵宿主后其生存环境发生骤变,菌体为适应变化的温度,碳源,偏酸或中性的PH值以及缺氧、高二氧化碳的生存环境不得不发生形态转换,转呈寄生状态生长,并形成致病性。大量研究证实处于信号转导通路上游位置的双组分信号系统在双相真菌适应体内的高渗透压及氧化刺激、形态转化、细胞壁生物合成以及毒力、致病性形成等方面发挥重要作用。双组分信号系统广泛存在于荚膜组织胞浆菌、皮炎芽生菌、马内菲青霉菌、白念珠菌等多种病原真菌体内,而人类细胞中尚未发现该信号转导系统的存在,因此,探明真菌双组分信号转导系统的机制可为抑制物的设计和寻找提供多个靶点,针对这些靶点研发的新型抗真菌药物将有效地抑制病原真菌的生长而不对宿主细胞造成损伤。近年,病原真菌双组份信号转导系统中组氨酸蛋白基因受到了学者的广泛关注,白念珠菌组氨酸蛋白激酶CaHK1/CaNIK1/CaCOS1/CaSLN1、粗糙脉胞霉菌组氨酸蛋白激酶NIK-1、皮炎芽生菌组氨酸蛋白酶DRK1、荚膜组织胞浆菌组氨酸蛋白酶DRK1相继被鉴定出来,并被证实为调控真菌致病性的形成及毒力蛋白表达的关键基因。
与其他双相真菌病相比,孢子丝菌病发病率最高,病原菌的毒力相对较弱,具有很好的实验安全性,并因大多仅感染皮肤和皮下组织而便于观察的特点可望成为双相真菌病防治研究的突破点。先前研究已经证实孢子丝菌双组份信号系统组氨酸蛋白激酶DRK1基因的存在,并对其进行克隆、结构功能域预测及致病性的鉴定。所以对其功能进行更深入的研究、并以其为靶点开发崭新的抗真菌药物及治疗方法,具有崭新的意义。
技术实现要素:
本发明的目的是构建一种孢子丝菌DRK1基因GFP报告菌株,以绿色荧光蛋白表达情况来示踪DRK1的表达水平,从而对DRK1基因的表达做到更加便捷、准确的监测。
本发明采用的技术方案是:孢子丝菌DRK1基因GFP报告菌株的构建方法,包括如下步骤:
(一)提取孢子丝菌总DNA;
(二)克隆孢子丝菌SsDRK1基因加尾信号序列至p-EGFP-N1载体的gfp基因尾部,制备重组质粒p-EGFP-N1-t:
2.1)以DNA为模板,以特异性引物Au和Ad进行PCR扩增,扩增具有孢子丝菌SsDRK1基因加尾信号序列的PCR产物;
所述的特异性引物Au:gcggcggccgcTAGGGACAAGGACCTCCAC
所述的特异性引物Ad:gcgtctagactagtGTAGTAAACAATCAAAACCAGTG
所述的孢子丝菌SsDRK1基因加尾信号序列的核苷酸序列如SEQ ID NO.1所示;
2.2)将PCR产物和p-EGFP-N1载体分别用NotI和XbaI双酶切,将酶切回收的PCR产物和p-EGFP-N1连接;
2.3)将连接产物转化大肠杆菌DH5a,获得重组质粒p-EGFP-N1-t;
(三)克隆孢子丝菌SsDRK1基因启动子序列至重组质粒p-EGFP-N1-t的gfp基因前部,制备重组质粒p-EGFP-N1-p-t:
3.1)以DNA为模板,以特异性引物Qu和Qd进行PCR扩增,扩增具有孢子丝菌SsDRK1基因启动子序列的PCR产物;
所述的特异性引物Qu:gcggagctcGCGAATGGACAGAGGTAAGT
所述的特异性引物Qd:gcgggatccTGTTGAAAGAGGGTAAACGGAG
所述的孢子丝菌SsDRK1基因启动子序列的核苷酸序列如SEQ ID NO.2所示;
3.2)将PCR产物和p-EGFP-N1-t分别用SacI和BamHI双酶切,将酶切回收的PCR产物和p-EGFP-N1-t连接;
3.3)将连接产物转化大肠杆菌DH5a,获得重组质粒p-EGFP-N1-p-t。
(四)构建PCB309-pSsDRK1-yEGFP-tSsDRK1重组质粒:
通过SacI和speI双酶切重组质粒p-EGFP-N1-p-t和载体PCB309,将双酶切后的产物进行连接,将重组质粒p-EGFP-N1-p-t上的“启动子-gfp-加尾信号”片段克隆至PCB309载体,构建PCB309-pSsDRK1-yEGFP-tSsDRK1重组质粒;
(五)构建孢子丝菌DRK1基因GFP报告菌株:
将PCB309-pSsDRK1-yEGFP-tSsDRK1重组质粒转化根癌农杆菌感受态细胞中,再将含有PCB309-pSsDRK1-yEGFP-tSsDRK1的根癌农杆菌与孢子丝菌混合,将PCB309-pSsDRK1-yEGFP-tSsDRK1转入孢子丝菌中,获得孢子丝菌DRK1基因GFP报告菌株。
本发明的有益效果是:
(1)应用范围:按照本发明的构建方法制备的孢子丝菌DRK1基因GFP报告菌株,可用于DRK1基因功能的研究,双组份信号通路上下游基因的鉴定,双相真菌病治疗方法的筛选。
(2)应用前景:本发明,方法操作简便,可通过基因工程的方法大量获得,具有很好的应用前景
(3)本发明的方法可成为基因功能分析的工具,并用于新型药物的研发及孢子丝菌病治疗方法的开发研究中。
附图说明
图1是孢子丝菌SsDRK1基因加尾信号序列的扩增产物双酶切鉴定结果。
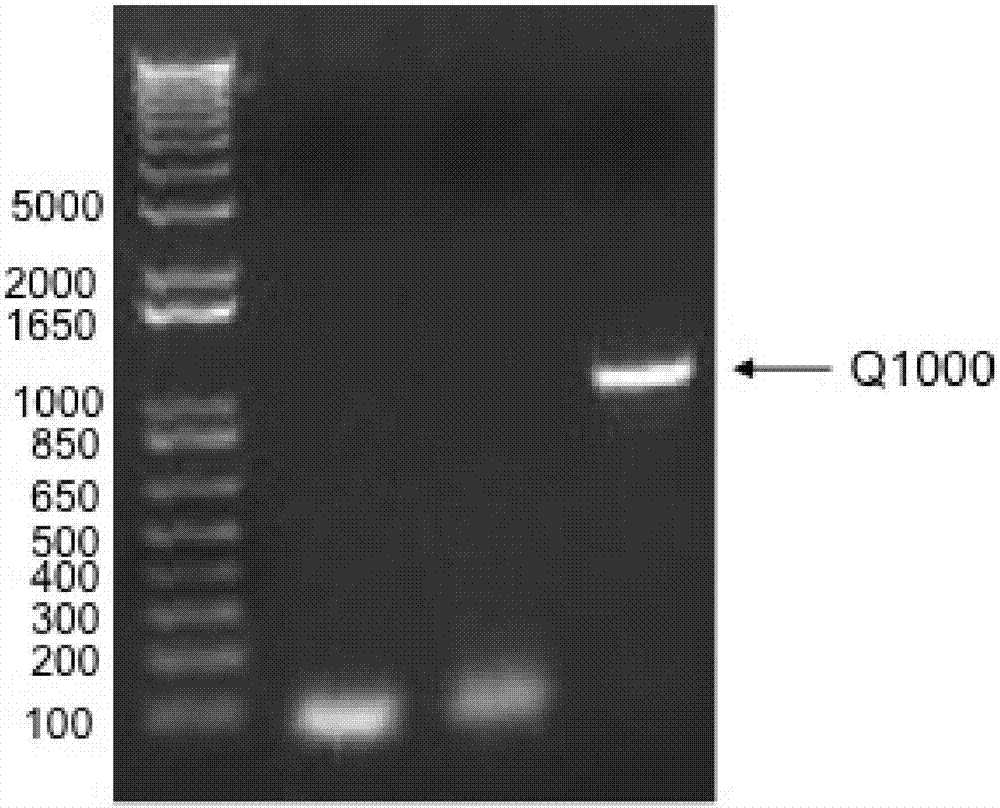
图2是孢子丝菌SsDRK1基因启动子序列的扩增产物双酶切鉴定结果。
图3是PCB309-pSsDRK1-yEGFP-tSsDRK1载体双酶切鉴定结果。
图4是不同孵育温度下孢子丝菌DRK1基因GFP报告菌株荧光表达情况;
其中,A,37°;B,40°;C,42°。
具体实施方式
实施例1 孢子丝菌DRK1基因GFP报告菌株的构建方法
(一)提取孢子丝菌总DNA;
(二)克隆孢子丝菌SsDRK1基因加尾信号序列至p-EGFP-N1载体的gfp基因尾部,制备重组质粒p-EGFP-N1-t:
2.1)以DNA为模板,以特异性引物Au和Ad进行PCR扩增,扩增具有孢子丝菌SsDRK1基因加尾信号序列的PCR产物;
所述的特异性引物Au:gcggcggccgcTAGGGACAAGGACCTCCAC
所述的特异性引物Ad:gcgtctagactagtGTAGTAAACAATCAAAACCAGTG
反应条件为:94℃预变性1min;94℃变性30sec,55℃退火15sec,72℃延伸1min,30个循环;72℃10min。
所述的孢子丝菌SsDRK1基因加尾信号序列的核苷酸序列如SEQ ID NO.1所示;
将PCR扩增产物进行凝胶电泳鉴定,结果如图1所示,由图1可见,产物大小与预期630bp相符。
2.2)将PCR产物和p-EGFP-N1载体分别用NotI和XbaI双酶切,将酶切回收的PCR产物和p-EGFP-N1连接;
酶切温度:37℃,时间:3h;
连接温度:22℃,时间:2h;
2.3)将连接产物转化大肠杆菌DH5a,获得重组质粒p-EGFP-N1-t;
将连接产物,筛选阳性克隆,经双酶切验证和由测序公司进行测序。经双酶切验证和测序分析综合表明,重组质粒p-EGFP-N1-t构建成功。
(三)克隆孢子丝菌SsDRK1基因启动子序列至重组质粒p-EGFP-N1-t的gfp基因前部,制备重组质粒p-EGFP-N1-p-t:
3.1)以DNA为模板,以特异性引物Qu和Qd进行PCR扩增,扩增具有孢子丝菌SsDRK1基因启动子序列的PCR产物;
所述的特异性引物Qu:gcggagctcGCGAATGGACAGAGGTAAGT
所述的特异性引物Qd:gcgggatccTGTTGAAAGAGGGTAAACGGAG
反应条件:94℃预变性1min;94℃变性30sec,55℃退火15sec,72℃延伸1min,30个循环;72℃10min。
所述的孢子丝菌SsDRK1基因启动子序列的核苷酸序列如SEQ ID NO.2所示;
将PCR扩增产物进行凝胶电泳鉴定,结果如图2所示,由图2可见,产物大小与预期1007bp相符
3.2)将PCR产物和p-EGFP-N1-t分别用SacI和BamHI双酶切,将酶切回收的PCR产物和p-EGFP-N1-t连接;
酶切温度:37℃,时间:3h;
连接温度:22℃,时间:2h。
3.3)将连接产物转化大肠杆菌DH5a,获得重组质粒p-EGFP-N1-p-t。
(四)构建PCB309-pSsDRK1-yEGFP-tSsDRK1重组质粒:
通过SacI和speI双酶切重组质粒p-EGFP-N1-p-t和PCB309载体,将双酶切后的产物进行连接,将重组质粒p-EGFP-N1-p-t上的“启动子-gfp-加尾信号”片段克隆至PCB309载体,构建PCB309-pSsDRK1-yEGFP-tSsDRK1重组质粒;
酶切温度:37℃,时间:3h;
连接温度:22℃,时间:2h。
(五)构建孢子丝菌DRK1基因GFP报告菌株:
将PCB309-pSsDRK1-yEGFP-tSsDRK1重组质粒转化根癌农杆菌感受态细胞中,再将含有PCB309-pSsDRK1-yEGFP-tSsDRK1的根癌农杆菌与孢子丝菌混合,将PCB309-pSsDRK1-yEGFP-tSsDRK1转入孢子丝菌中,获得孢子丝菌DRK1基因GFP报告菌株。
5.1)制备根癌农杆菌感受态细胞
5.2)以PCB309-pSsDRK1-yEGFP-tSsDRK1重组质粒及空转质粒PCB309转化根癌农杆菌感受态细胞,挑选阳性克隆验证;
将质粒采用电击的方法转化根癌农杆菌,将感受态细胞和待转化的DNA于冰上操作,取待转化的质粒PCB309-pSsDRK1-yEGFP-tSsDRK1和PCB309各1μl混入感受态细胞中,混匀。
吸取置于已经预冷的电击杯中,擦去电击杯外的冷凝水和蒸汽,推入电击仪中,电压为1.8V,在电击杯中加入1mlLB培养基,冲洗,使转化后的细胞与培养基充分混合,吸取入试管中,28℃培养活化1小时。
将培养好的菌液加入EP管中离心,弃上清,用剩余的液体重悬菌体,全部涂到含有抗生素Kan的LB培养基中。
培养2天后挑取阳性克隆,提取PCB309-pSsDRK1-yEGFP-tSsDRK1质粒,验证。
将PCB309-pSsDRK1-yEGFP-tSsDRK1重组质粒,筛选阳性克隆,经双酶切验证,结果如图3所示,由图3可见酶切后两片段均与预期大小相符,由测序公司进行测序。双酶切验证和测序分析综合表明,PCB309-pSsDRK1-yEGFP-tSsDRK1重组质粒构建成功。
5.3)根癌农杆菌介导重组载体PCB309-pSsDRK1-yEGFP-tSsDRK1转染孢子丝菌
制备申克氏孢子丝菌ATCC10268分生孢子和农杆菌细胞。将农杆菌与申克氏孢子丝菌共培养。
筛选转化子:将制得的共培养的含有农杆菌和分生孢子的菌落转移到筛选培养基平板(诱导培养基平板中加入100mg/L潮霉素,200uM头孢噻肟钠)上培养4天,收集生长的阳性菌落接种于沙氏培养基,4℃保存,得到孢子丝菌DRK1基因GFP报告菌株。
实施例2 孢子丝菌DRK1基因GFP报告菌株感受环境温度的确认
方法:改变孢子丝菌培养环境的温度,荧光显微镜观察绿色荧光的强弱来示踪孢子丝菌DRK1基因表达水平的变化,将于37°BHI培养基中培养获得的酵母相DRK1-GFP报告菌株置于40°和42°孵箱中继续培养,荧光显微镜观察DRK1的表达,结果图如4所示,由图4可见随着温度的升高,荧光亮度发生改变。
<110> 香港大学深圳医院
<120> 孢子丝菌DRK1基因GFP报告菌株的构建方法
<160> 2
<210> 1
<211> 630
<212> DNA
<210> 孢子丝菌SsDRK1基因加尾信号序列
<400> 1
tagggacaag gacctccact tgtttccctt cctcctccct ctgtcttgtg ttctcaaaac 60
aactggatat gctaacattt cattttcttg tacagcttct tcgcgcgcat agtagctaag 120
agaggccctc acatacaacg tttacctacg atgttttcaa ctaagctacc cctccatggc 180
gttgcactac gaacaactca tctttctatt ttgtgtttta tggcttttta tctggcaccc 240
cttgggtgac atactcacgt cgattttggg ctatcggtat ttctgggctt catatttctt 300
gttgaagcgt gtttctttcc atcatgtgga ggggagagaa ggggaacgag gagatcaaga 360
caacgtcttt ttttgtctac tcgccctctg gggacttcaa ggaagcgaga catgatcgca 420
tcatatttgg cgtttacgat attgtttgtc ttttttggct tttatctttt gacgtttttc 480
atgctgttga gatttgaatc tctgcgtctc gtatctcatg ttatgtcctg tttgtttatt 540
tcctctctac gacgtataat atgtgccaag tcaccattgg ccccgcgacg gcttttcaat 600
ttcatttcac tggttttgat tgtttactac 630
<210> 2
<211> 1007
<212> DNA
<210> 孢子丝菌SsDRK1基因启动子序列
<400> 1
gcgaatggac agaggtaagt tgtgtatggg aggcggcctt gtcctgttgc tttcctcaac 60
cttcccttgg ctacttgacg cgcctgacga gaccattcct tagtccttga ggcccctgtt 120
cttgcccacc gccactgact ctcccgaagg ctcccactat ctgccaccat ctcccaaccg 180
tctctcccta gctgtccctt tcccctcccc tggagcctgt gcccatccgc ctgcagatac 240
cctgaaagca tccactttcc cccaccctat cgacctgtcc ccccctcttg tcgccattct 300
ggcgcctcat ctggtccctt gtccttcgtt gtccgccccc tacttccttg tactttccca 360
tgctcattta gctaaccgta tcgattccca tccagccttg gcagcggtaa caagcctaag 420
aattgctggc ggcacgcaag cctcccgcta ccatttgcct ttttcccacc atccctctcc 480
acctgctcgc ctcactctca gcatccctac cttttttttc tcatatccag cattttcatt 540
tctcgtatcc atcccctctt gtcctgtctc ccctttccca gctcctttta actttctgat 600
cgagttttct ttacagcgaa tcgaactgta tcatcgccat caccaactgg cgacgaaata 660
cgtcaagcag cacccgctgg cagttcaaga gtcccctttt tgttgccatc tctcttaccg 720
ccccaaagcg tccctttttg ccgccttttc cgctcatcgc aaccacgaac cattgagcgc 780
gttgtcacga cccaaaaggc gacttctccc cgtttggggg atcgccacgc aacaaccgga 840
cacttgttga gaaccttaaa tttcacaaaa gagctacttt aacccgctct aagacatttg 900
ctttcgccat ccggccgtgt ttcccgagtc aaggcgaaag cttctacacg cagcgcgtcc 960
caatctcagc tttcgcccct gttacctccg tttaccctct ttcaaca 1007
- 还没有人留言评论。精彩留言会获得点赞!