一种芳烃钌配合物及其制备方法与应用与流程
本发明涉及合成化学技术领域,尤其是涉及一种芳烃钌配合物,本发明还涉及一种芳烃钌配合物的制备方法与应用。
背景技术:
芳烃钌配合物具有经典的“钢琴凳”空间结构,是一类以强疏水性的苯或者取代苯作为辅助配体,通过苯环上π轨道上的电子与中心金属钌形成共价键的小分子金属有机化合物。从1967年,winkhaus和singer首次合成芳烃钌(ii)化合物[1]开始,国内外就有大量文献报道各种不同结构类型的芳烃钌配合物的合成和抗肿瘤的作用。
kondapi等人报道以[(η6-c6h6)ru(dmso)cl2]2+为代表的芳烃钌配合物对人结肠癌colo-205细胞和乳腺癌zr-75-1细胞均表现出良好的抑制作用,同时对dna具有很强的亲和力,还能抑制拓扑异构酶ii来抑制肿瘤细胞的生长与增殖,显示此类配合物的特性[]vashishtgyn,konurun,kondapiak.topoisomeraseiiantagonismandanticanceractivityofcoor-dinatedderivativesof[rucl2(c6h6)(dmso)][j].arch.biochem.biophys,2002,401(1):53-62]。
dyson等人报道的一类以金刚烷胺为配体的芳烃钌(ii)配合物是通过[rucl2(η6-arene)]2(0.64mmol)和pta(1.28mmol)在甲醇溶液中加热回流24小时得到,产率85-95%。[scolaroc,bergamoa,brescacinl,delfinor,cocchiettom,laurenczyg,geldbachtj,savag,dysonpj.invitroandinvivoevaluationofruthenium(ii)-areneptacomplexes.[j].jmedchem,2005,48:4161–4171]此类配合物对ts/a老鼠腺癌细胞有一定活性(ic5060~300μm),对人体正常细胞毒性较小(ic50>300μm)。其中rapta-b([ru(η6-c6h6)(pta)cl2])和rapta-c([ru(η6-p-c6h4meipr)(pta)cl2])通过调控线粒体和p53–jnk多种细胞信号通路,调高p21的水平和减少细胞周期蛋白e的量,导致肿瘤细胞发生g2/m期阻滞,继而诱导细胞凋亡。[chatterjees,kundus,bhattacharyyaa,hartingercg,dysonpj,theruthenium(ii)–arenecompoundrapta-cinducesapoptosisineaccellsthroughmitochondrialandp53–jnkpathways.[j].jbiolinorg-chem,2008,13:1149–1155]体内实验发现rapta-c能在患有mca乳腺癌的cba老鼠体内通过抑制血管生成来抑制肿瘤增殖和转移。[nowak-sliwinskap,vanbjr,casinia.organometal-licruthnium(ii)arenecompoundswithantiangiogenicactivity[j].j.med.chem.2011,54(11):3895-3902]
sadler等人报道的乙二胺配位的芳烃钌(ii)配合物是通过[rucl2(η6-arene)]2(0.64mmol)和乙二胺衍生物(2.0mmol)在甲醇溶剂中搅拌1.5小时,后加入nh4pf6,过滤,取滤液于冰箱中重结晶6小时得到。产率37.7%。这类化合物在体外实验中对a2780人卵巢癌有很好活性,但没有交叉耐药性。该化合物可以紧密结合dna的鸟嘌呤,是抗肿瘤细胞活性实现靶向定位的重要途径。[morrisre,airdre,delsocorromurdochp,chenh,cummingsj,hughesnd,parsonss,parkina,boydg,jodrelldi,sadlerpj.inhibitionofcancercellgrowthbyruthenium(ii)arenecomplexes.jmedchem,2001,44:3616–3621]其中rm175([(η6-biphenyl)ru(ethylenediamine)-cl]+)通过促进细胞-细胞再粘连和降低金属蛋白酶(mmps)的释放来抑制肿瘤的侵袭与转移。[bergamoa,masia,peacockaf,habtemariama,sadlerpj,savagj.invivotumourandmetastasisreductionandinvitroeffectsoninvasionassaysoftherutheniumrm175andosmiumafap51organometallicsinthemammarycancermodelinorg.biochem,2010,104:79-86,[habtemariama,melchartm,fernandezr,parsonss,oswaldidh,parkina,fabbianifpa,davidsonje,dawsona,airdre,jodrelldi,sadlerpj.struc-ture-activityrelationshipsforcytotoxicruthenium(ii)arenecomplexescontainingn,n-,n,o-,ando,o-chelatingligands.j.medchem,2006,49:6858–6868]sadler等人报道的2,4-戊二酮配位的芳烃钌(ii)配合物[(η6-arene)ru(β-diketonate)cl]通过[rucl2(η6-arene)]和乙酰丙酮钠在丙酮中搅拌50min得到,产率61.2%。这类配合物在体外实验中对人卵巢癌a2780细胞的ic50值取决于芳环辅助配体的种类,在同类配体下,芳环为对甲基异丙基苯活性最高,[(η6-p-cymene)ru(β-diketonate)cl]对人卵巢癌细胞a2780的ic50为17μm,具有良好的抗肿瘤作用。
国内毛宗万教授等人报道的咔啉配位的芳烃钌(ii)配合物是通过[rucl2(η6-arene)]0mmol)和咔啉衍生物(2.00mmol)在甲醇溶液中回流2.5小时得到。产率78%。此类配合物在体外的抗肿瘤活性比卡铂类高3~12倍,没有交叉耐药性,对正常细胞毒性小,对顺铂耐药的细胞a549cisr具有较高活性,对正常人非成纤维细胞hlf具有较低毒性。该类配合物通过下调cdk1和cyclinb1的表达,直接靶向cdk1(细胞周期素依赖性激酶),导致肿瘤细胞发生g2m期阻滞,还能够通过跟线粒体有关的通路诱导肿瘤细胞凋亡并能提高细胞内活性氧物质ros的量。[hel,liaosy,tancp,yerr,xuyw,zhaom,,jiln,maozw.ruthenium–arene–β-carbolinecomplexesaspotentinhibitorsofcyclin-dependentkinase1:synthesis,characterizationandanticancermechanismstudies.chem.eur.j,2013,19,12152–12160
国内苏炜教授等人报导的缩氨基硫脲化合物配位的3个芳烃钌(ii)配合物[(η6-p-cymene)ru(r-bztsc)cl]cl(bztsc=苯甲醛缩氨基硫脲,r=h,ch3andc6h5)是通过[rucl2(η6-p-cymene)](0.05mmol)和苯甲醛缩氨基硫脲(0.1mmol)在丙酮溶液中45℃加热回流6小时得到。产率39%。此类芳烃钌(ii)配合物在体外实验中对鼻咽癌、肺癌、乳腺癌和卵巢癌细胞均有很好的抗增殖活性。其中配合物[(η6-p-cymene)ru(c6h5-bztsc)cl]cl的活性最好,ic50值分别为20μm,31μm,10μm和34μm[suw,zhouq,huangym,etal.synthesis,crystalandelectronicstructure,anticanceractivityofruthenium(ii)arenecomplexeswiththiosemicarbazones[j].appl.organometal.chem.2013,27(5):307-312.]。
目前的芳烃钌配合物,根据配体的不同,通常针对不同的肿瘤的作用不同,因此,对于不同的肿瘤症状,需要研发出不同的芳烃钌配合物。
技术实现要素:
本发明的目的之一,在于提出一种芳烃钌配合物,对多种肿瘤株具有高抑制作用。
为解决上述技术问题,本发明采用以下技术方案予以实现:一种芳烃钌配合物,采用rpip(pip:2-苯基咪唑[4,5f][1,10]菲啰啉)为主配体,结构式如下:
其中,所述r独立的选自氢,碳原子数为1~6的烷基或碳原子为1~6的取代烷基,苯基或取代苯基,吡啶基或取代吡啶基,呋喃基或取代呋喃基,吡咯基或取代吡咯基,噻唑或取代噻唑基。前述苯基、吡啶基、呋喃基、噻唑的取代基均选自羟基、硝基、卤素、氨基、羧基、氰基、巯基、碳原子数为3~8的环烷基、so3h、碳原子数为1~6的烷基、碳原子数为2~6的链烯基、碳原子为2~6的链炔基、羟基(c1-c6)烷基、氨基(c1-c6)烷基、co2r’、conr’r’、cor’、so2r’r’、(c1-c6)烷氧基、(c1-c6)烷硫基、-n=nr’、nr’r’或三氟(c1-c6)烷基中的任一种;其中,所述r’选自h、碳原子数为1~6的烷基或苯基。
进一步,所述芳烃钌配合物的化学式为[(η6-ch3c6h5)ru(rpip)cl]y。
其中,所述y选自氯离子、碘离子、溴离子、高氟酸根、3-氟合磷酸根,磺酸根中任一种,优选氯离子。
作为优选的,所述芳烃钌配合物的结构式为
上述结构的物质,能够抑制肿瘤细胞的增殖,对肺腺癌细胞,成骨肉瘤细胞、鼻咽癌和高转移性乳腺癌细胞生长等均具有很好的抑制作用;而对人体正常细胞毒性较小。
如其中的化学式为[ru(ch3c6h5)(m-clpip)cl]cl,缩写为ram052的所述芳烃钌配合物,结构式如下:
,其能选择性抑制mda-mb-231乳腺癌细胞,ic50为3.22μm;对c6脑胶质瘤细胞,ic50为3.8μm;对cne-1鼻咽癌细胞,ic50为26.44μm;对人体正常细胞hk-2毒性小,ic50为434.5μm。通过光谱学和粘度、热变性实验结果表明,芳烃钌配合物能够以插入结合的方式与dna分子缔合,对细胞dna的功能产生微扰,并最终诱导肿瘤细胞凋亡。申请人对其更详细的机制研究发现,[(η6-c6h6)ru(o-clpip)cl]cl通过下调cyclina和cdk2蛋白水平,最终使肿瘤瘤细胞mg-63抑制在s期。彗星实验表明[(η6-c6h6)ru(o-clpip)cl]cl通过上调p-p53和p-histoneh2a表达水平造成mg-63细胞dna损伤。芳烃钌配合物可能是通过介导p53磷酸化过程来诱导肿瘤细胞dna损伤,导致肿瘤细胞发生s期阻滞,最终死亡。体外dna结合实验发现该化合物能稳定c-mycg-四链体dna结构,下调c-myc蛋白的表达,从而抑制肿瘤细胞的增殖。
又比如,所述芳烃钌配合物的化学式为[ru(ch3c6h5)(p-npip)cl]cl,缩写为ram143,
其对不同肿瘤细胞株都表现出适当的抑制作用,特别是mda-mb-231乳腺癌细胞,ic50为21.69μm;对c6脑胶质瘤细胞,ic50为7.00μm;对cne-1鼻咽癌细胞,ic50为99.7μm;对人体正常细胞hacat毒性小。
又比如,所述芳烃钌配合物的化学式为[ru(ch3c6h5)(o-mpip)cl]cl,缩写为ram271,
,其对不同肿瘤细胞株都表现出适当的抑制作用,mda-mb-231乳腺癌细胞,ic50为24.24μm;对c6脑胶质瘤细胞,ic50为21.39μm;对人体正常细胞hk-2毒性小,ic50为191.7μm。
本发明的另一目的在于提出一种芳烃钌配合物的制备方法,工艺简单,易于推广。
所述芳烃钌配合物的制备方法,所述芳烃钌配合物的制备微波合成法。
进一步,所述芳烃钌配合物的制备方法,由[(η6-ch3c6h5)rucl]2cl2,通过1-甲基-1,4-环己二烯衍生物和菲并咪唑衍生物在二氯甲烷溶剂中,30-180℃下,微波辐射反应得到。
进一步,所述芳烃钌配合物的制备方法,反应的温度为90℃,时间为15s~60min。
本发明的另一目的在于提出一种芳烃钌配合物的应用,所述芳烃钌配合物在抑制肿瘤中的应用。更进一步的,所述芳烃钌配合物在抑制乳腺癌、脑胶质瘤、白血病、鼻咽癌和肺腺癌中的应用。
附图说明
图1为ram051的esi-ms图谱;
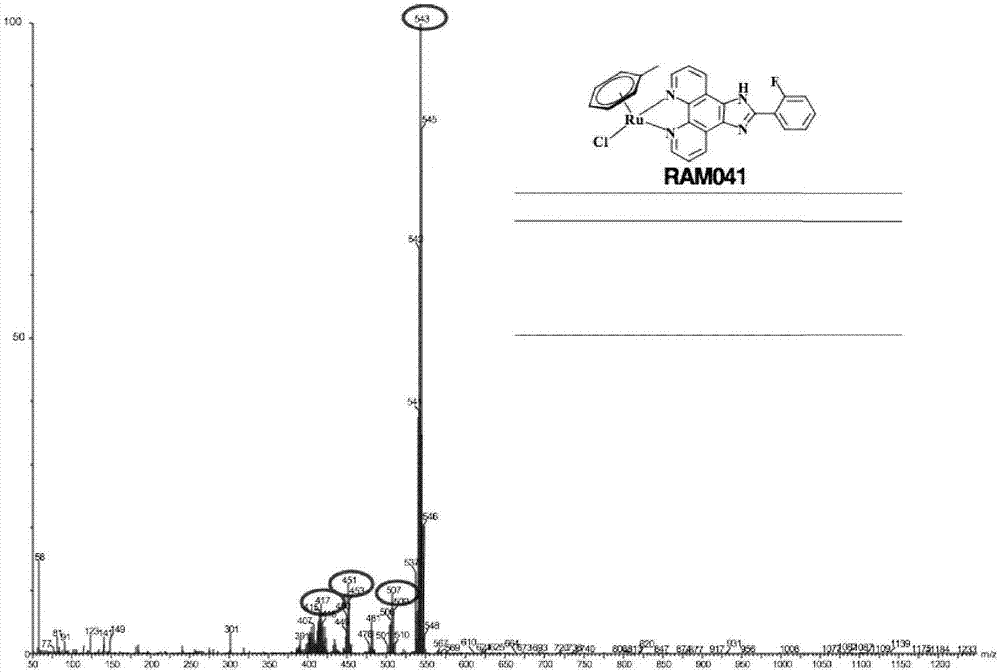
图2为ram041的esi-ms图谱;
图3为ram01的esi-ms图谱;
图4为ram09的esi-ms图谱;
图5为ram042的esi-ms图谱;
图6为ram073的esi-ms图谱;
图7为ram081的esi-ms图谱;
图8为ram211的esi-ms图谱;
图9为ram203的esi-ms图谱;
图10为ram201的esi-ms图谱;
图11为ram093的esi-ms图谱;
图12为ram141的esi-ms图谱;
图13为ram143的esi-ms图谱;
图14为ram15的esi-ms图谱;
图15为ram271的esi-ms图谱;
图16为ram13的esi-ms图谱;
图17为ram103的esi-ms图谱;
图18为ram082的esi-ms图谱;
图19为ram031的esi-ms图谱;
图20为ram032的esi-ms图谱;
图21为ram052的esi-ms图谱;
图22为ram061的esi-ms图谱;
图23为ram062的esi-ms图谱。
具体实施方式
为让本领域的技术人员更加清晰直观的了解本发明,下面将结合附图,对本发明作进一步的说明。
实施例1
ram01的微波合成
由rucl3与1-甲基-1,4-环己二烯反应30-180℃下,微波辐射15s-60min得到中间体,由前述反应得到的中间体和咪唑[4,5f][1,10]菲啰啉(ip)在二氯甲烷溶剂中,60℃下,微波辐射15s。反应结束后,用少量甲醇溶解,得到ram01,结构式如下。
实施例2
ram09的微波合成
前述步骤同实施例1;
中间体和2-苯基-咪唑[4,5f][1,10]菲啰啉(pip)在二氯甲烷溶剂中,120℃下,微波辐射30min,得到ram09,结构式如下。
实施例3
ram031的微波合成
前述步骤同实施例1;
中间体和2-(2-氟代苯基)-咪唑[4,5f][1,10]菲啰啉(pip)在二氯甲烷溶剂中,90℃下,微波辐射30min。
实施例4
ram032的微波合成
前述步骤同实施例1;
中间体和2-(3-氟代苯基)-咪唑[4,5f][1,10]菲啰啉(o-fpip)在二氯甲烷溶剂中,90℃下,微波辐射30min。
实施例5
ram051的微波合成
前述步骤同实施例1;
中间体和2-(2-氯代苯基)-咪唑[4,5f][1,10]菲啰啉(o-clpip)在二氯甲烷溶剂中,90℃下,微波辐射30min。
实施例6
ram052的微波合成
前述步骤同实施例1;
中间体和2-(3-氯代苯基)-咪唑[4,5f][1,10]菲啰啉(m-clpip)在二氯甲烷溶剂中,90℃下,微波辐射30min。
实施例7
ram061的微波合成
前述步骤同实施例1;
中间体和2-(2-溴代苯基)-咪唑[4,5f][1,10]菲啰啉(o-brpip)在二氯甲烷溶剂中,90℃下,微波辐射30min。
实施例8
ram031的微波合成
前述步骤同实施例1;
中间体和2-(2-羟基苯基)-咪唑[4,5f][1,10]菲啰啉(o-hpip)在二氯甲烷溶剂中,90℃下,微波辐射30min。
实施例9
ram032的微波合成
前述步骤同实施例1;
中间体和2-(3-羟基苯基)-咪唑[4,5f][1,10]菲啰啉(m-hpip)在二氯甲烷溶剂中,90℃下,微波辐射30min。
实施例10
ram081的微波合成
前述步骤同实施例1;
中间体和2-(2-三氟甲苯基)-咪唑[4,5f][1,10]菲啰啉(o-tfmpip)在二氯甲烷溶剂中,90℃下,微波辐射30min。
实施例11
ram082的微波合成
前述步骤同实施例1;
中间体和2-(3-三氟甲苯基)-咪唑[4,5f][1,10]菲啰啉(m-tfmpip)在二氯甲烷溶剂中,90℃下,微波辐射30min。
实施例12
ram211的微波合成
前述步骤同实施例1;
中间体和2-(2-羧基苯基)-咪唑[4,5f][1,10]菲啰啉(o-cpip)在二氯甲烷溶剂中,90℃下,微波辐射30min。
实施例13
ram213的微波合成
前述步骤同实施例1;
中间体和2-(4-羧基苯基)-咪唑[4,5f][1,10]菲啰啉(p-cpip)在二氯甲烷溶剂中,90℃下,微波辐射30min。
实施例14
ram13的微波合成
前述步骤同实施例1;
中间体和2-(4-乙炔苯基)-咪唑[4,5f][1,10]菲啰啉(p-epip)在二氯甲烷溶剂中,90℃下,微波辐射30min。
实施例15
ram201的微波合成
前述步骤同实施例1;
中间体和2-(2-甲氧苯基)-咪唑[4,5f][1,10]菲啰啉(o-mopip)在二氯甲烷溶剂中,90℃下,微波辐射30min。
实施例16
ram203的微波合成
前述步骤同实施例1;
中间体和2-(4-甲氧苯基)-咪唑[4,5f][1,10]菲啰啉(p-mopip)在二氯甲烷溶剂中,90℃下,微波辐射30min。
实施例17
ram073的微波合成
前述步骤同实施例1;
中间体和2-(4-碘代苯基)-咪唑[4,5f][1,10]菲啰啉(p-ipip)在二氯甲烷溶剂中,90℃下,微波辐射30min。
实施例18
ram103的微波合成
前述步骤同实施例1;
中间体和2-(4-磺酸苯基)-咪唑[4,5f][1,10]菲啰啉(p-spip)在二氯甲烷溶剂中,90℃下,微波辐射30min。
实施例19
ram093的微波合成
前述步骤同实施例1;
中间体和2-(4-n,n’-二甲胺苯基)-咪唑[4,5f][1,10]菲啰啉(p-dmapip)在二氯甲烷溶剂中,90℃下,微波辐射30min。
实施例20
ram141的微波合成
前述步骤同实施例1;
中间体和2-(2-硝基苯基)-咪唑[4,5f][1,10]菲啰啉(o-npip)在二氯甲烷溶剂中,90℃下,微波辐射30min。
实施例21
ram143的微波合成
前述步骤同实施例1;
中间体和2-(4-硝基苯基)-咪唑[4,5f][1,10]菲啰啉(p-npip)在二氯甲烷溶剂中,90℃下,微波辐射30min。
实施例22
ram271的微波合成
前述步骤同实施例1;
中间体和2-(2-甲苯基)-咪唑[4,5f][1,10]菲啰啉(o-mpip)在二氯甲烷溶剂中,90℃下,微波辐射30min。
实施例23ram15
实施例24
rpip为配体的芳烃钌配合物对不同肿瘤细胞的抗肿瘤活性的研究
结果如下表:
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种荔枝多糖铁配合物的制备方法
- 一种含4,4’-二溴-2,2’-联吡啶的新型钌配合物的制备方法及其抗肿瘤活性的制作方法
- 一种羰基锝-99m标记的美法仑配合物及其制备方法与用图
- 一种锝-99m标记的美法仑配合物及其制备方法与用图
- Ru配合物Ru(bpy)<sub>3</sub>(NH<sub>2</sub>)<sub>2</sub>与石墨烯量子相结合的方法
- 1,4-二甲基-2,5-二-1h-1,2,4-双三唑二维铜配合物单晶与应用
- 1,4-二甲基-2,5-二-1h-双三唑均苯四酸钴配合物单晶与应用
- 一种含双氮配体低价金属铑(Ⅰ)的环辛二烯配合物及其制备方法
- 1,4-二甲基-2,5-二-1h-双三唑三维铜配合物单晶与应用
- 一种2-羰基-3-苯基丙酸苯甲酰腙二(2,4-二氯苄基)锡配合物及其制备方法和应用