一种2,4‑二氯‑5‑氟苯甲酰氯的制备方法与流程
技术领域:
本发明属于有机合成领域,涉及制备氟喹诺酮类中间体2,4-二氯-5-氟苯甲酰氯的方法。
背景技术:
:
喹诺酮类是人工合成的含4-喹诺酮母核为基本结构的抗菌药物,从20世纪70年代后期诺氟沙星问世以来,第三代喹诺酮类-氟喹诺酮类药物的研究和开发引起了抗菌药物的革命,出现了很多有临床价值的新药,如氧氟沙星、环丙沙星、罗美沙星、氟罗沙星等,成为临床主要的抗感染药物之一,仅次于头孢菌类和青霉素类药物。氟喹诺酮类药物的结构特点是在6-位有氟原子,在7-位有取代氨基,文献报道的合成路线很多,但这些原料来源困难,且价格高。
大部分是从氟氯苯乙酮(2,4-二氯-5-氟苯乙酮)为起始原料,与碳酸二乙酯缩合,再与原甲酸三乙酯进行乙氧亚甲基化,不同胺胺化,最后经环合和不同的哌嗪取代完成。该路线主要缺点是路线中两步使用了活性高的钠氢,存在安全隐患。
目前用2,4-二氯氟苯作为原料,制备2,4-二氯-5-氟苯甲酰氯中间体,与不同的胺基丙烯酸甲酯缩合,经环合、取代得到氟喹诺酮类,此合成路线较短,安全性高,操作简单。
随着沙星类药物需求量不断夸大,其合成中的中间体2,4-二氯-5-氟苯甲酰氯的需求量也日益增加,关于它的合成方法也很多,安永彬等人以2,4-二氯氟苯等为原料,以三氯化铝为催化剂,naclo溶液为氧化剂,在70-80℃反应,总收率为78.4%,该方法使用次氯酸钠等高污染、高风险的原料,且次氯酸钠需大大过量,污染大、成本高(中国抗生素杂质29(2004):529-530)。
温新民等人提出了使用2,4-二氯氟苯与乙酰氯在三氯化铝的存在下反应制备2,4-二氯-5-氟苯乙酮,然后在硝酸作用氧化成酸,经过酰化的2,4-二氯-5-氟苯甲酰氯,该制备方法氧化反应需要过来的硝酸,造成成本的浪费,且酰化反应需要绝对无水,不适合放大生产(济宁医学院学报23(2000):21-22)。
selsaku.k等人使用2,4-二氯氟苯在alcl3催化下与四氯化碳反应引入三氯甲基,经酸性水解得到2,4-二氯-5-氟苯甲酸,再与二氯亚砜酰化得到2,4-二氯-5-氟苯甲酰氯。该制备方法第一步有约20%-30%的副产物(fcl2c6h2)2ccl2,使得原料的利用低,造成成本高,难以实现工业化生产(us5241111)。
为了降低2,4-二氯氟苯与四氯化碳反应时产生二聚物的比例,吴政杰等人发明了固体酸催化剂s2o82-/sm2o3-zro2-al2o3以及李乙刚等人发明了复合固体超强酸催化剂,在这些催化剂的作用下,生成2,4-二氯-5-氟-(三氯甲基)苯,再经fecl3催化水解即得。该方法虽然降低了二聚物的比例,但无法避免二聚物的生成;并且合成催化剂步骤繁琐,成本高,且活化温度高达到600℃,存在安全隐患,不适合放大生产(cn104725221,cn104649890)。
技术实现要素:
:
为克服现有技术中的上述原料利用率低,有副产物二聚物产生的缺点,本发明提供了一种将二聚物继续转化合成2,4-二氯-5-苯甲酰氯的方法,使得原料转化率80%以上,改变了目前原料来源困难,利用率低的缺点,节约资源,降低了生产成本,并且操作简单,易于放大生产。
本发明提供了一种合成氟喹诺酮类药物关键中间体(ⅱ)即2,4-二氯-5-氟苯甲酰氯的方法,采用2,4-二氯氟苯为原料,经过傅克反应、水解后生成中间体2,4-二氯-5-氟苯甲酰氯,并且将反应产生的副产物即二聚物(ⅲ)经过水解、氧化和酰化转化为最终产物化合物(ⅱ),总收率达到88%以上。
本发明提供了一种合成2,4-二氯-5-氟苯甲酰氯的合成方法,所述方法包括以下步骤:
(1)傅克反应:将2,4-二氯氟苯在路易斯酸三氯化铝的作用下和四氯化碳反应,得到中间体(ⅰ)2,4-二氯-5-氟三氯甲苯;
(2)水解反应:中间体(ⅰ)在三氯化铁的作用水解得到中间体(ⅱ);
本发明提供了一种合成2,4-二氯-5-氟苯甲酰氯的合成方法,所述方法包括以下步骤:
(3)水解反应:中间体(ⅲ)加热至熔融状态,加入催化量三氯化铁,缓慢滴加水,反应得到中间体(ⅳ);
(4)氧化反应:中间体(ⅳ)在20℃-40℃下,与氧化剂反应得到中间体(ⅴ)
(5)酰化反应:中间体(ⅴ)与二氯亚砜在50-100℃下进行酰化反应,得到2,4-二氯-5-氟苯甲酰氯。
步骤(1)中,2,4-二氯氟苯与四氯化碳的摩尔比为:1:0.95-1:1.0;温度为0℃-70℃,优选40℃-70℃;路易斯酸为三氯化铁和三氯化铝中的一种;
步骤(2)中,三氯化铁与中间体(ⅰ)的质量比为1:20-1:10,中间体(ⅰ)与水的摩尔比为1:0.95-1:1.0.
步骤(3)中,温度为80℃-120℃,温度过高,会损失加入的水,从而导致收率降低,优选90℃-100℃,三氯化铁与中间体(ⅲ)的质量比为1:100-1:20,中间体(ⅲ)与水的摩尔比为1:1.0-1:1.1.
步骤(4)中的氧化剂有常见的具有强氧化性酸:浓硫酸和硝酸,考虑到硝酸的安全性问题,优选浓硫酸;具有氧化性的盐,高锰酸钾、重铬酸钾、高氯酸钾,优选成本低、污染小的高锰酸钾;过氧化物氧化剂:双氧水、次氯酸钠等,优选双氧水,后处理操作简单,收率高;在强氧化剂中,采用的溶剂为大极性溶剂,丙酮、n,n-二甲基甲酰胺、1,4-二氧六环等,中性氧化剂可以选择醇类作为溶剂,甲醇、乙醇或者异丙醇等。
步骤(5)中,该反应采用无溶剂反应,中间体(ⅴ)与二氯亚砜的摩尔比为1:1.0-1:1.1,引发剂为n,n-二甲基甲酰胺,温度0℃-100℃。
有益效果
1、采用简单易制备的方法合成出关键中间体(ⅱ),避免使用高污染、高风险原料,未使用任何破坏性溶剂,污染物排放少;
2、第一步副产物重新利用转化为产物,使得原料的转化率升至为80%以上,突破了卤代芳烃与四氯化碳反应副产物转化的技术瓶颈;
3、本发明的工艺路线简洁,原料易得,成本低,反应条件温和,操作简便,制备出的2,4-二氯-5-氟苯甲酰氯纯度高且收率高达88%以上。
说明书附图
图1为实施例1中的化合物i的1hnmr谱图
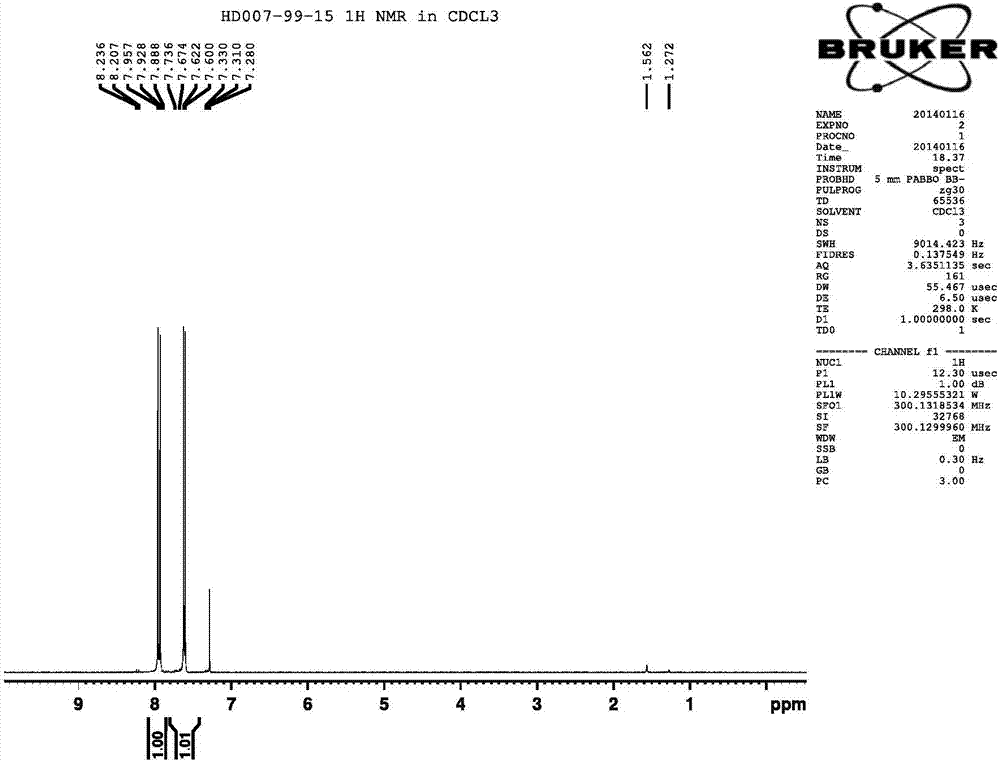
图2为实施例1中的化合物ii的1hnmr谱图
图3为实施例1中的化合物ⅴ的1hnmr谱图
具体实施方式
实施例1:一种2,4-二氯-5-氟苯甲酰氯的制备方法,总收率为89.3%.
将原料2,4-二氯氟苯(100g,0.606mol)加入到四氯化碳(107.4g,0.667mol)中,搅拌溶解后,加入三氯化铁(1.52g,9.3mmol),升温至70℃并维持2小时,液相检测原料基本消失。降至室温,加入2.5mhclaq(500ml)淬灭反应,搅拌至澄清后分层,取下层有机相减压蒸馏,得化合物ⅰ(128.5g,75.1%)和釜残即化合物ⅲ(25.3g,20.3%)。
1hnmr(cdcl3300mhz):δ7.548-7.646(d,1h),8.024-8.115(d,1h).
将无水三氯化铁(1g)加入到化合物ⅰ(20g,70.9mmol)中,加热至145℃后,缓慢滴加水(1.27g,70.9mmol),加完继续搅拌30分钟,气相检测原料消失,减压蒸馏得化合物ⅱ(15.8g,98.1%)
1hnmr(cdcl3300mhz):δ7.600-7.736(d,1h),7.688-7.957(d,1h).
向第一步的釜残即化合物ⅲ(17g,39.7mmol)加入fecl3(0.17g)、h2o(0.75g,41.7mmol),加热至120℃,搅拌至无气体产生,反应结束。将反应液冷却至室温,缓慢滴入冰水中(250ml),边滴加边搅拌,有白色析出,过滤烘干的化合物ⅳ(14.5g,98%)。
1hnmr(cdcl3300mhz):δ7.718-7.723(d,2h),7.914-7.918(d,2h).
化合物ⅳ(10g,26.8mmol)加入乙醇(30ml)中,搅拌澄清后,加入30%双氧水(5ml,44.1mmol),加热回流过夜,反应完全。加入100ml水,6n盐酸水溶液调至ph=1-2,有大量固体析出,过滤烘干的化合物ⅴ(4.5g,80.3%)
1hnmr(cdcl3300mhz):δ7.831-7.862(d,1h),7.921-7.943(d,1h).
于反应瓶中加入化合物ⅴ(65.3g,0.312mol),二氯亚砜(40g,0.336mol)和1滴dmf,加热至80℃,搅拌1小时,液相检测反应结束。常压蒸馏除去二氯亚砜,减压蒸出产品的化合物ⅱ(69.5,98%)。
实施例2:一种2,4-二氯-5-氟苯甲酰氯的制备方法,总收率为92.1%
步骤(1):步骤与实施例1相同,所不同的地方在于步骤(1),本实施例的步骤(1)为将原料2,4-二氯氟苯(10g,60.6mmol)加入到四氯化碳(9.27g,57.6mmol)中,搅拌溶解后,加入三氯化氯(0.124g,0.93mmol),升温至40℃并维持2小时,液相检测原料基本消失。降至室温,加入2.5mhclaq(50ml)淬灭反应,搅拌至澄清后分层,取下层有机相减压蒸馏,得化合物ⅰ(13.1g,76.6%)和釜残即化合物ⅲ(2.8g,22.6%)
步骤(2):步骤与实施例1相同,将无水三氯化铁(2g)加入到化合物ⅰ(40g,141.8mmol)中,加热至140℃后,缓慢滴加水(2.54g,141.8mmol),加完继续搅拌30分钟,气相检测原料消失,减压蒸馏得化合物ⅱ(31.9g,99%)。
步骤(3):步骤与实施例1相同,向第一步的釜残即化合物ⅲ(17g,39.7mmol)加入fecl3(0.17g)、h2o(0.75g,41.7mmol),加热至120℃,搅拌至无气体产生,反应结束。将反应液冷却至室温,缓慢滴入冰水中(250ml),边滴加边搅拌,有白色析出,过滤烘干的化合物ⅳ(14.5g,98%)。
步骤(4):步骤与实施例1相同,所不同的地方在于步骤(4),化合物ⅳ(10g,26.8mmol)加入丙酮(30ml)中,搅拌澄清后,加入高锰酸钾(8.47g,53.6mmol),加热回流过夜,反应完全。加入100ml水,6n盐酸水溶液调至ph=1-2,有大量固体析出,过滤烘干的化合物ⅴ(4.3g,76.7%)
步骤(5):步骤与实施例1相同,于反应瓶中加入化合物ⅴ(120g,0.574mol),二氯亚砜(73.6g,0.618mol)和1滴dmf,加热至100℃,搅拌2小时,液相检测反应结束。常压蒸馏除去二氯亚砜,减压蒸出产品的化合物ⅱ(125.3g,96.1%)。
实施例3:一种2,4-二氯-5-氟苯甲酰氯的制备方法,总收率为88.5%
步骤(1):与实施例1相同,将原料2,4-二氯氟苯(100g,0.606mol)加入到四氯化碳(107.4g,0.667mol)中,搅拌溶解后,加入三氯化铁(1.52g,9.3mmol),升温至70℃并维持2小时,液相检测原料基本消失。降至室温,加入2.5mhclaq(500ml)淬灭反应,搅拌至澄清后分层,取下层有机相减压蒸馏,得化合物ⅰ(128.5g,75.1%)和釜残即化合物ⅲ(25.3g,20.3%)。
步骤(2):与实施例1相同,所不同的地方在于步骤(2),本实施例的步骤(2)将无水三氯化铁(3g)加入到化合物ⅰ(30g,106.3mmol)中,加热至145℃后,缓慢滴加水(1.52g,101.0mmol),加完继续搅拌30分钟,气相检测原料消失,减压蒸馏得化合物ⅱ(22.96g,95.2%)。
步骤(3):步骤与实施例1相同,向第一步的釜残即化合物ⅲ(17g,39.7mmol)加入fecl3(0.17g)、h2o(0.75g,41.7mmol),加热至120℃,搅拌至无气体产生,反应结束。将反应液冷却至室温,缓慢滴入冰水中(250ml),边滴加边搅拌,有白色析出,过滤烘干的化合物ⅳ(14.5g,98%)。
步骤(4):步骤与实施例1相同,所不同的地方在于步骤(4),化合物ⅳ(50g,134mmol)加入丙酮(150ml)中,搅拌澄清后,加入高锰酸钾(268g,53.6mmol),加热回流过夜,反应完全。加入500ml水,6n盐酸水溶液调至ph=1-2,有大量固体析出,过滤烘干的化合物ⅴ(24.9g,88.8%)
步骤(5):步骤与实施例1相同,于反应瓶中加入化合物ⅴ(120g,0.574mol),二氯亚砜(73.6g,0.618mol)和1滴dmf,加热至100℃,搅拌2小时,液相检测反应结束。常压蒸馏除去二氯亚砜,减压蒸出产品的化合物ⅱ(125.3g,96.1%)。
实施例4:一种2,4-二氯-5-氟苯甲酰氯的制备方法,总收率为89.6%
步骤(1):将原料2,4-二氯氟苯(100g,0.606mol)加入到四氯化碳(107.4g,0.667mol)中,搅拌溶解后,加入三氯化铁(1.52g,9.3mmol),升温至70℃并维持2小时,液相检测原料基本消失。降至室温,加入2.5mhclaq(500ml)淬灭反应,搅拌至澄清后分层,取下层有机相减压蒸馏,得化合物ⅰ(128.5g,75.1%)和釜残即化合物ⅲ(25.3g,20.3%)
步骤(2):将无水三氯化铁(2g)加入到化合物ⅰ(40g,141.8mmol)中,加热至140℃后,缓慢滴加水(2.54g,141.8mmol),加完继续搅拌30分钟,气相检测原料消失,减压蒸馏得化合物ⅱ(31.9g,99%)。
步骤(3):步骤与实施例1相同,所不同的地方在于步骤(3),本实施例的步骤(3)向第一步的釜残即化合物ⅲ(25g,58.4mmol)加入fecl3(1.25g)、h2o(1.155g,64.2mmol),加热至120℃,搅拌至无气体产生,反应结束。将反应液冷却至室温,缓慢滴入冰水中(350ml),边滴加边搅拌,有白色析出,过滤烘干的化合物ⅳ(21.2g,97.8%)。
步骤(4):步骤与实施例1相同,所不同的地方在于步骤(4),本实施例的步骤(4),化合物ⅳ(20g,53.6mmol)加入浓硫酸(50ml)中,加热至90℃,搅拌澄清后,即反应完全。加入100ml水,有大量固体析出,过滤烘干的化合物ⅴ(8.9g,80.1%)。
步骤(5):于反应瓶中加入化合物ⅴ(120g,0.574mol),二氯亚砜(73.6g,0.618mol)和1滴dmf,加热至100℃,搅拌2小时,液相检测反应结束。常压蒸馏除去二氯亚砜,减压蒸出产品的化合物ⅱ(125.3g,96.1%)
实施例5:一种2,4-二氯-5-氟苯甲酰氯的制备方法,总收率为92.1%.
步骤(1):步骤与实施例1相同,所不同的地方在于步骤(1),本实施例的步骤(1)为将原料2,4-二氯氟苯(10g,60.6mmol)加入到四氯化碳(9.27g,57.6mmol)中,搅拌溶解后,加入三氯化氯(0.124g,0.93mmol),升温至40℃并维持2小时,液相检测原料基本消失。降至室温,加入2.5mhclaq(50ml)淬灭反应,搅拌至澄清后分层,取下层有机相减压蒸馏,得化合物ⅰ(13.1g,76.6%)和釜残即化合物ⅲ(2.8g,22.6%)
步骤(2):步骤与实施例1相同,将无水三氯化铁(2g)加入到化合物ⅰ(40g,141.8mmol)中,加热至140℃后,缓慢滴加水(2.54g,141.8mmol),加完继续搅拌30分钟,气相检测原料消失,减压蒸馏得化合物ⅱ(31.9g,99%)。
步骤(3):步骤与实施例1相同,所不同的地方在于步骤(3),本实施例的步骤(3)向第一步的釜残即化合物ⅲ(21.5g,50.2mmol)加入fecl3(2.15g)、h2o(0.9g,50.2mmol),加热至90℃,搅拌至无气体产生,反应结束。将反应液冷却至室温,缓慢滴入冰水中(300ml),边滴加边搅拌,有白色析出,过滤烘干的化合物ⅳ(18.3g,98.1%)。
步骤(4):步骤与实施例1相同,所不同的地方在于步骤(4),化合物ⅳ(10g,26.8mmol)加入丙酮(30ml)中,搅拌澄清后,加入高锰酸钾(8.47g,53.6mmol),加热回流过夜,反应完全。加入100ml水,6n盐酸水溶液调至ph=1-2,有大量固体析出,过滤烘干的化合物ⅴ(4.3g,76.7%)
步骤(5):步骤与实施例1相同,于反应瓶中加入化合物ⅴ(120g,0.574mol),二氯亚砜(73.6g,0.618mol)和1滴dmf,加热至100℃,搅拌2小时,液相检测反应结束。常压蒸馏除去二氯亚砜,减压蒸出产品的化合物ⅱ(125.3g,96.1%)。
实施例6:一种2,4-二氯-5-氟苯甲酰氯的制备方法,总收率为88.7%。
步骤(1):与实施例1相同,将原料2,4-二氯氟苯(100g,0.606mol)加入到四氯化碳(107.4g,0.667mol)中,搅拌溶解后,加入三氯化铁(1.52g,9.3mmol),升温至70℃并维持2小时,液相检测原料基本消失。降至室温,加入2.5mhclaq(500ml)淬灭反应,搅拌至澄清后分层,取下层有机相减压蒸馏,得化合物ⅰ(128.5g,75.1%)和釜残即化合物ⅲ(25.3g,20.3%)。
步骤(2):与实施例1相同,所不同的地方在于步骤(2),本实施例的步骤(2)将无水三氯化铁(3g)加入到化合物ⅰ(30g,106.3mmol)中,加热至145℃后,缓慢滴加水(1.52g,101.0mmol),加完继续搅拌30分钟,气相检测原料消失,减压蒸馏得化合物ⅱ(22.96g,95.2%)。
步骤(3):步骤与实施例1相同,向第一步的釜残即化合物ⅲ(17g,39.7mmol)加入fecl3(0.17g)、h2o(0.75g,41.7mmol),加热至120℃,搅拌至无气体产生,反应结束。将反应液冷却至室温,缓慢滴入冰水中(250ml),边滴加边搅拌,有白色析出,过滤烘干的化合物ⅳ(14.5g,98%)。
步骤(4):步骤与实施例1相同,所不同的地方在于步骤(4),化合物ⅳ(10g,26.8mmol)加入dmf(30ml)中,搅拌澄清后,加入高氯酸钾(7.4g,53.6mmol),加热回流过夜,反应完全。加入100ml水,6n盐酸水溶液调至ph=1-2,有大量固体析出,过滤烘干的化合物ⅴ(5.1g,91%)。
步骤与实施例1相同,所不同的地方在于步骤(5),本实施例的步骤(5)于反应瓶中加入化合物ⅴ(100g,0.478mol),二氯亚砜(56.9g,0.478mol)和1滴dmf,加热至80℃,搅拌1小时,液相检测反应结束。常压蒸馏除去二氯亚砜,减压蒸出产品的化合物ⅱ(103.5g,95.2%)。
实施例7:一种2,4-二氯-5-氟苯甲酰氯的制备方法,总收率为88.6%。
步骤(1):将原料2,4-二氯氟苯(100g,0.606mol)加入到四氯化碳(107.4g,0.667mol)中,搅拌溶解后,加入三氯化铁(1.52g,9.3mmol),升温至70℃并维持2小时,液相检测原料基本消失。降至室温,加入2.5mhclaq(500ml)淬灭反应,搅拌至澄清后分层,取下层有机相减压蒸馏,得化合物ⅰ(128.5g,75.1%)和釜残即化合物ⅲ(25.3g,20.3%)
步骤(2):将无水三氯化铁(2g)加入到化合物ⅰ(40g,141.8mmol)中,加热至140℃后,缓慢滴加水(2.54g,141.8mmol),加完继续搅拌30分钟,气相检测原料消失,减压蒸馏得化合物ⅱ(31.9g,99%)。
步骤(3):步骤与实施例1相同,所不同的地方在于步骤(3),本实施例的步骤(3)向第一步的釜残即化合物ⅲ(25g,58.4mmol)加入fecl3(1.25g)、h2o(1.155g,64.2mmol),加热至120℃,搅拌至无气体产生,反应结束。将反应液冷却至室温,缓慢滴入冰水中(350ml),边滴加边搅拌,有白色析出,过滤烘干的化合物ⅳ(21.2g,97.8%)。
步骤(4):步骤与实施例1相同,所不同的地方在于步骤(4),本实施例的步骤(4),化合物ⅳ(20g,53.6mmol)加入浓硫酸(50ml)中,加热至90℃,搅拌澄清后,即反应完全。加入100ml水,有大量固体析出,过滤烘干的化合物ⅴ(8.9g,80.1%)。
步骤(5):步骤与实施例1相同,所不同的地方在于步骤(5),本实施例的步骤(5)于反应瓶中加入化合物ⅴ(20g,95.6mmol),二氯亚砜(11.38g,100.4mmol)和1滴dmf,于冰浴下反应1小时后升至室温搅拌,继续搅拌1小时,液相检测反应结束。常压蒸馏除去二氯亚砜,减压蒸出产品的化合物ⅱ(19.5g,90%)
实施例8:一种2,4-二氯-5-氟苯甲酰氯的制备方法,总收率为94.8%。
步骤(1):步骤与实施例1相同,所不同的地方在于步骤(1),本实施例的步骤(1)为将原料2,4-二氯氟苯(10g,60.6mmol)加入到四氯化碳(9.27g,57.6mmol)中,搅拌溶解后,加入三氯化氯(0.124g,0.93mmol),升温至40℃并维持2小时,液相检测原料基本消失。降至室温,加入2.5mhclaq(50ml)淬灭反应,搅拌至澄清后分层,取下层有机相减压蒸馏,得化合物ⅰ(13.1g,76.6%)和釜残即化合物ⅲ(2.8g,22.6%)
步骤(2):步骤与实施例1相同,将无水三氯化铁(2g)加入到化合物ⅰ(40g,141.8mmol)中,加热至140℃后,缓慢滴加水(2.54g,141.8mmol),加完继续搅拌30分钟,气相检测原料消失,减压蒸馏得化合物ⅱ(31.9g,99%)。
步骤(3):步骤与实施例1相同,向第一步的釜残即化合物ⅲ(17g,39.7mmol)加入fecl3(0.17g)、h2o(0.75g,41.7mmol),加热至120℃,搅拌至无气体产生,反应结束。将反应液冷却至室温,缓慢滴入冰水中(250ml),边滴加边搅拌,有白色析出,过滤烘干的化合物ⅳ(14.5g,98%)。
步骤(4):步骤与实施例1相同,所不同的地方在于步骤(4),化合物ⅳ(10g,26.8mmol)加入丙酮(30ml)中,搅拌澄清后,加入重铬酸钾(15.7g,53.6mmol),加热回流过夜,反应完全。加入100ml水,6n盐酸水溶液调至ph=1-2,有大量固体析出,过滤烘干的化合物ⅴ(5.0g,89.1%)
步骤(5):步骤与实施例1相同,于反应瓶中加入化合物ⅴ(120g,0.574mol),二氯亚砜(73.6g,0.618mol)和1滴dmf,加热至100℃,搅拌2小时,液相检测反应结束。常压蒸馏除去二氯亚砜,减压蒸出产品的化合物ⅱ(125.3g,96.1%)。
实施例9:一种2,4-二氯-5-氟苯甲酰氯的制备方法,总收率为87.7%。
步骤(1):与实施例1相同,将原料2,4-二氯氟苯(100g,0.606mol)加入到四氯化碳(107.4g,0.667mol)中,搅拌溶解后,加入三氯化铁(1.52g,9.3mmol),升温至70℃并维持2小时,液相检测原料基本消失。降至室温,加入2.5mhclaq(500ml)淬灭反应,搅拌至澄清后分层,取下层有机相减压蒸馏,得化合物ⅰ(128.5g,75.1%)和釜残即化合物ⅲ(25.3g,20.3%)。
步骤(2):与实施例1相同,所不同的地方在于步骤(2),本实施例的步骤(2)将无水三氯化铁(3g)加入到化合物ⅰ(30g,106.3mmol)中,加热至145℃后,缓慢滴加水(1.52g,101.0mmol),加完继续搅拌30分钟,气相检测原料消失,减压蒸馏得化合物ⅱ(22.96g,95.2%)。
步骤(3):步骤与实施例1相同,向第一步的釜残即化合物ⅲ(17g,39.7mmol)加入fecl3(0.17g)、h2o(0.75g,41.7mmol),加热至120℃,搅拌至无气体产生,反应结束。将反应液冷却至室温,缓慢滴入冰水中(250ml),边滴加边搅拌,有白色析出,过滤烘干的化合物ⅳ(14.5g,98%)。
步骤(4):步骤与实施例1相同,所不同的地方在于步骤(4),本实施例的步骤(4),化合物ⅳ(10g,26.8mmol)加入丙酮(30ml)中,搅拌澄清后,加入高锰酸钾(8.47g,53.6mmol),加热回流过夜,反应完全。加入100ml水,6n盐酸水溶液调至ph=1-2,有大量固体析出,过滤烘干的化合物ⅴ(4.8g,85.6%)
步骤与实施例1相同,所不同的地方在于步骤(5),本实施例的步骤(5)于反应瓶中加入化合物ⅴ(100g,0.478mol),二氯亚砜(56.9g,0.478mol)和1滴dmf,加热至80℃,搅拌1小时,液相检测反应结束。常压蒸馏除去二氯亚砜,减压蒸出产品的化合物ⅱ(103.5g,95.2%)。
实施例10:一种2,4-二氯-5-氟苯甲酰氯的制备方法,总收率为88.5%。
步骤(1):将原料2,4-二氯氟苯(100g,0.606mol)加入到四氯化碳(107.4g,0.667mol)中,搅拌溶解后,加入三氯化铁(1.52g,9.3mmol),升温至70℃并维持2小时,液相检测原料基本消失。降至室温,加入2.5mhclaq(500ml)淬灭反应,搅拌至澄清后分层,取下层有机相减压蒸馏,得化合物ⅰ(128.5g,75.1%)和釜残即化合物ⅲ(25.3g,20.3%)
步骤(2):将无水三氯化铁(2g)加入到化合物ⅰ(40g,141.8mmol)中,加热至140℃后,缓慢滴加水(2.54g,141.8mmol),加完继续搅拌30分钟,气相检测原料消失,减压蒸馏得化合物ⅱ(31.9g,99%)。
步骤(3):步骤与实施例1相同,所不同的地方在于步骤(3),本实施例的步骤(3)向第一步的釜残即化合物ⅲ(25g,58.4mmol)加入fecl3(1.25g)、h2o(1.155g,64.2mmol),加热至120℃,搅拌至无气体产生,反应结束。将反应液冷却至室温,缓慢滴入冰水中(350ml),边滴加边搅拌,有白色析出,过滤烘干的化合物ⅳ(21.2g,97.8%)。
步骤与实施例1相同,所不同的地方在于步骤(4),本实施例的步骤(4),化合物ⅳ(20g,53.6mmol)加入浓硫酸(50ml)中,加热至90℃,搅拌澄清后,即反应完全。加入100ml水,有大量固体析出,过滤烘干的化合物ⅴ(8.9g,80.1%)。
步骤(5):步骤与实施例1相同,所不同的地方在于步骤(5),本实施例的步骤(5)于反应瓶中加入化合物ⅴ(20g,95.6mmol),二氯亚砜(11.38g,100.4mmol)和1滴dmf,于冰浴下反应1小时后升至室温搅拌,继续搅拌1小时,液相检测反应结束。常压蒸馏除去二氯亚砜,减压蒸出产品的化合物ⅱ(19.5g,90%)。
- 还没有人留言评论。精彩留言会获得点赞!