超支化聚氧乙烯醚及其聚合制备的侧链超支化聚羧酸的制作方法
本发明涉及一种超支化聚氧乙烯醚及其制备方法、以及以其为大单体,聚合制备的侧链超支化聚羧酸及其制备方法,属于有机合成化学及精细化学品技术领域。
背景技术:
聚羧酸类化合物是通过丙烯酸、丙烯酸酯和酸酐类的小单体和聚氧乙烯醚大单体在引发剂作用下共聚获得的化合物。目前,传统梳形结构聚羧酸主要被分为四大类:(1)甲基丙烯/烯酸甲酯共聚型聚羧酸;(2)烯丙基醚类共聚型聚羧酸;(3)酰胺/酰亚胺共聚型聚羧酸;(4)两性共聚型聚羧酸。四种不同类型聚羧酸的分类方法主要以主链和侧链的连接方式来区分,无论哪一种类型的传统聚羧酸,侧链均为直链聚氧乙烯醚。
传统梳形结构聚羧酸的直链聚氧乙烯醚所能提供的氢键作用、亲水性、表面活性、引气性有限。
技术实现要素:
本发明要解决的技术问题是:提高传统聚羧酸的侧链密度,降低传统聚羧酸的表面张力和表观粘度,提高传统聚羧酸的起泡性能。本发明通过提高传统聚羧酸的侧链密度,降低传统聚羧酸的表面张力和表观粘度,提高传统聚羧酸的起泡性能。
为了解决上述技术问题,本发明合成的侧链超支化聚羧酸,采用超支化聚氧乙烯醚替代传统直链聚氧乙烯醚作为合成用大单体。可增加聚羧酸侧链密度,在相同乙氧基聚合度下,增加聚氧乙烯醚大单体的乙氧基密度,可降低聚羧酸溶液的表面张力和表观黏度,有效提高聚羧酸的起泡性能。
本发明提供一种超支化聚氧乙烯醚,具有如式1所示结构:
a-(b)2m-1-(d)x
式1
其中,a为具有如下结构的基团:
b为具有如下结构的基团:
d为具有如下结构的基团:
式1中,m为大于等于0的整数;
当m为0时,x为1,a基团与d基团中的n连接,通式如下:
当m为1时,x为2,a基团连接b基团中的n,b基团的亚甲基连接d基团的n,通式如下:
当m大于等于2时,x为2m,a基团连接b基团中的n,b基团的亚甲基再连接下一个b基团的n,位于最后端的b基团的亚甲基连接d基团的n,通式如下:
n为5-100的整数。
本发明还提供一种侧链超支化聚羧酸,所述侧链超支化聚羧酸由含不饱和双键的小单体与权利要求1所述的超支化聚氧乙烯醚中的一种或多种聚合得到,其中,所述含不饱和双键的小单体为:丙烯酸、丙烯酸酯、马来酸酐中的一种或多种。
其中,所述侧链超支化聚羧酸的结构通式如下式所示:
式中,b基团为具有如下结构的基团:
d基团为具有如下结构的基团:
r为c1~c5的直链烷基,n=5-100,a,b,c为0~200之间的整数,且a,b,c不同时为0,d为20~200之间的整数。
本发明还提供上述的超支化聚氧乙烯醚的制备方法,m为0时,该超支化聚氧乙烯醚的制备方法包括以下步骤:
(1)甲氧基聚乙二醇(mpeg)和碳酸钾,共同放入有机溶剂中,同时将氯化亚砜溶于有机溶剂中,回流条件下将氯化亚砜溶液滴加到mpeg中,反应完全,而后过滤除盐,减压蒸馏,得到cl-mpeg;
(2)将cl-mpeg溶于有机溶剂中,加入碳酸钾、碘化钾以及乙醇胺,黑暗条件氮气保护回流直至反应完全,而后砂芯漏斗过滤,减压蒸馏,得到二臂接枝产物;
(3)将二臂接枝产物、丙烯酰氯溶于有机溶剂,并在二臂接枝产物溶液中加入碳酸钾,将丙烯酰氯溶液冰浴条件下滴加到二臂接枝产物混合液中,而后黑暗氮气保护反应,过滤后减压蒸馏,得到二臂超支化聚氧乙烯醚。
m为1时,该超支化聚氧乙烯醚的制备方法包括以下步骤:
(1)mpeg和碳酸钾,共同放入有机溶剂中,同时将氯化亚砜溶于有机溶剂中,回流条件下将氯化亚砜溶液滴加到mpeg溶液中,反应完全,而后过滤除盐,减压蒸馏,得到cl-mpeg;
(2)将乙醇胺、丙烯酸甲酯分别溶于有机溶剂中,冰浴氮气保护条件下,将溶有丙烯酸甲酯溶液滴加完毕,而后继续冰浴反应0.5-2h,升温至20-40℃黑暗条件下反应至完全,减压蒸馏,得到树状小分子m0.5;
(3)取m0.5、乙二胺分别溶于溶剂中,冰浴氮气保护条件下向m0.5溶液中滴加乙二胺溶液,继续冰浴反应,升温至20-40℃而后在氮气保护且黑暗条件下继续反应至完全,减压蒸馏,得到树状小分子m1;
(4)取cl-mpeg,溶于有机溶剂中,加入碳酸钾、碘化钾和m1,黑暗条件、氮气保护回流进行反应,而后过滤,减压蒸馏,得到四臂接枝产物;
(5)将四臂接枝产物、丙烯酰氯溶于有机溶剂,并在四臂接枝产物溶液中加入碳酸钾,将丙烯酰氯溶液冰浴条件下滴加到四臂接枝产物混合液中,而后黑暗氮气保护反应,过滤后减压蒸馏,得到四臂超支化聚氧乙烯醚。
m大于等于2时,该超支化聚氧乙烯醚的制备方法包括以下步骤:
(1)mpeg和碳酸钾,共同放入有机溶剂中,同时将氯化亚砜溶于有机溶剂中,回流条件下将氯化亚砜溶液滴加到mpeg溶液中,在反应完全,而后过滤除盐,减压蒸馏,得到cl-mpeg;
(2)将乙醇胺、丙烯酸甲酯分别溶于有机溶剂中,冰浴氮气保护条件下,将溶有丙烯酸甲酯溶液滴加完毕,而后继续冰浴反应1-2h,升温至20-40℃在黑暗条件下反应至完全,减压蒸馏,得到树状小分子m0.5;
(3)取m0.5、乙二胺分别溶于溶剂中,冰浴氮气保护条件下向m0.5溶液中滴加乙二胺溶液,继续冰浴反应,升温至20-40℃后在氮气保护且黑暗条件下继续反应至完全,减压蒸馏,得到树状小分子m1;
(4)用制备出的不同代的树状小分子代替乙醇胺,重复步骤(2)-(3)直至制备出需要的m代树状小分子mm;
(5)取cl-mpeg,溶于有机溶剂中,加入碳酸钾、碘化钾和mm,黑暗条件、氮气保护回流进行反应,而后过滤,减压蒸馏,得到2(m+1)臂接枝产物;
(6)将2(m+1)臂接枝产物、丙烯酰氯溶于有机溶剂,并在四臂接枝产物溶液中加入碳酸钾,将丙烯酰氯溶液冰浴条件下滴加到四臂接枝产物混合液中,而后黑暗氮气保护反应,过滤后减压蒸馏,得到2(m+1)臂超支化聚氧乙烯醚。
本发明提供上述的侧链超支化聚羧酸的制备方法,包括如下步骤:
将所述超支化聚氧乙烯醚,溶于去离子水中,分别将含不饱和双键的小单体和链转移剂溶于去离子水中,引发剂溶于去离子水中,然后均滴加到超支化聚氧乙烯醚作为大单体的溶液中,滴完后保温反应,反应结束后冷却至室温,调节溶液ph至7-8,即得。
所述引发剂为无机过氧化物,引发剂的用量为大单体和小单体总质量的1%-10%。所述的链转移剂为:甲基丙烯磺酸钠、巯基丙酸、巯基乙酸中的一种或多种。
所述超支化型聚氧乙烯醚和含不饱和双键的小单体的摩尔比为1:(1~5)。
含不饱和双键的小单体和链转移剂溶于去离子水中,引发剂溶于去离子水中,60℃下均滴加到超支化聚氧乙烯醚的溶液中,滴完后保温反应3-6h。
本发明合成的侧链超支化聚羧酸,采用超支化聚氧乙烯醚替代传统直链聚氧乙烯醚作为合成用大单体。可增加聚羧酸侧链密度,在相同乙氧基聚合度下,增加聚氧乙烯醚大单体的乙氧基密度,可降低聚羧酸溶液的表面张力和表观黏度,有效提高聚羧酸的起泡性能。
附图说明
图1为本发明实施例中制备的cl-mpeg600的ftir谱图;
图2为本发明实施例中制备的cl-mpeg600的1hnmr谱图;
图3为本发明实施例中制备的cl-mpeg600的13cnmr谱图;
图4为本发明实施例中制备的m0.5的ftir谱图;
图5为本发明实施例中制备的m0.5的1hnmr谱图;
图6为本发明实施例中制备的m0.5的13cnmr谱图;
图7为本发明实施例中制备的m1的ftir谱图;
图8为本发明实施例中制备的m1的1hnmr谱图;
图9为本发明实施例中制备的m1的13cnmr谱图;
图10为本发明实施例中制备的以分子量为1000的mpeg为原料得到的二臂超支化聚氧乙烯醚的ftir谱图;
图11为本发明实施例中制备的四臂超支化聚氧乙烯醚的ftir谱图。
图12为本发明实施例中制备的实施例1的二臂超支化聚羧酸(1)的ftir谱图。
图13为本发明实施例中制备的实施例2的四臂超支化聚羧酸(1)的ftir谱图。
具体实施方式
下面结合附图和具体实施例对本发明作进一步说明,以使本领域的技术人员可以更好的理解本发明并能予以实施,但所举实施例不作为对本发明的限定。
实施例1
所述的侧链超支化聚羧酸,以丙烯酸为小单体,以甲基丙烯磺酸钠为链转移剂,以过硫酸铵为引发剂,以甲氧基封端的二臂超支化聚氧乙烯醚为大单体,通过水溶液自由基共聚反应得到二臂超支化聚羧酸。
其制备方法如下:
(1)分别于四个三口烧瓶中取400(n=8)、600(n=13)、1000(n=22)、2000(n=45)四种不同分子量的mpeg0.1mol。
mpeg的通式:
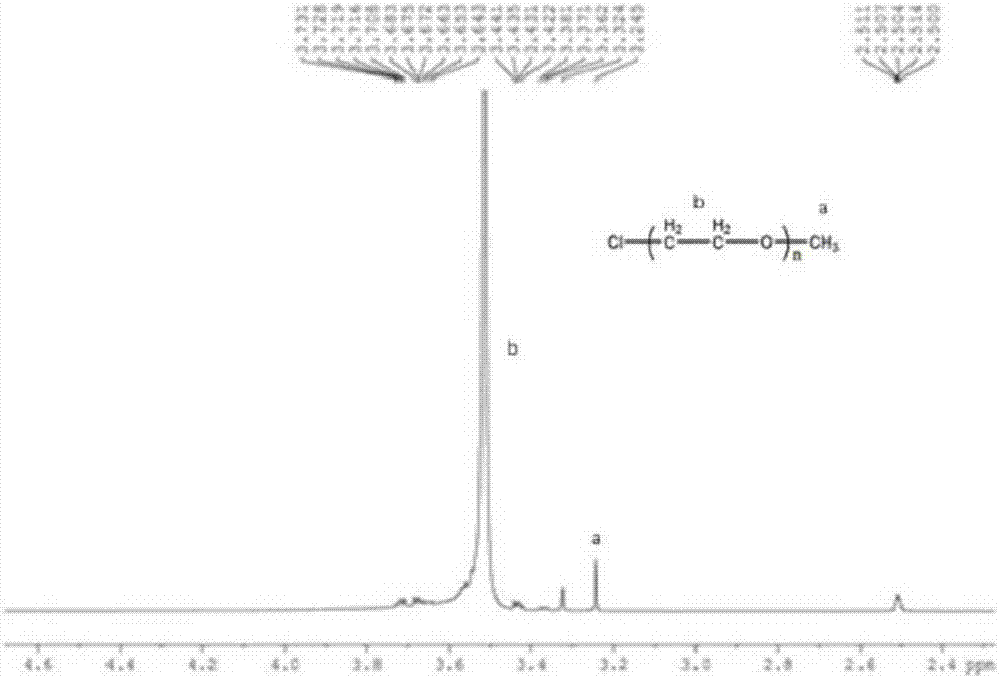
0.2mol碳酸钾,放入100ml二氯甲烷中,回流条件下滴加0.5mol氯化亚砜和30ml二氯甲烷的混合液,45℃条件下反应至完全,结束后用少量二氯甲烷冲洗回流冷凝管,并用砂芯漏斗过滤除盐,再减压蒸馏,得到四种分子量的cl-mpeg;cl-mpeg(分子量600的mpeg)的ftir谱图、1hnmr谱图和13cnmr谱图见图1~图3。图1中,2889cm-1处为-ch2-的对称伸缩振动峰,在1471cm-1处为-ch2-的变形振动峰,在1111cm-1处为c-o-c伸缩振动峰,在1365cm-1处为-ch3的对称变形振动峰,在663cm-1处为c-cl的伸缩振动峰。图2中,在3.32-3.73ppm出现多重峰,这是-ch2ch2o-重复单元中的-ch2-质子峰。在3.24ppm处的信号峰为末端-o-ch3中的质子峰。图3中,在70.14ppm处为-ch2ch2o-重复单元的-ch2-的碳原子信号峰,58.51ppm为末端-och3-上碳原子的化学位移,44.04ppm处出现的信号峰为c-cl中碳原子的信号峰。
(2)分别取上述四种不同分子量的cl-mpeg0.1mol,放入100ml乙腈中,加入0.2mol碳酸钾和0.8g碘化钾和0.04mol乙醇胺85℃,黑暗条件下氮气保护回流24h,而后乙腈清洗回流冷凝管,砂芯漏斗过滤,减压蒸馏,得到四种二臂接枝产物。
(3)将四种二臂接枝产物分别各自加入80ml乙腈中,并再加入0.2mol碳酸钾,将丙烯酰氯0.12mol溶于20ml乙腈中,置于恒压滴液漏斗中,冰浴滴加0.5h,而后黑暗氮气保护在室温下反应24h,过滤后减压蒸馏,即得四种甲氧基封端的二臂超支化聚氧乙烯醚大单体。以分子量为1000的mpeg为原料得到的甲氧基封端的二臂超支化聚氧乙烯醚大单体ftir谱图见图10,2888.53cm-1处为-ch2-的对称伸缩振动峰;1724.03cm-1处为酯的羰基峰(c=o);1647.80cm-1处为c=c的伸缩振动峰;1468.46cm-1处为-ch2-的变形振动峰;1344.85cm-1处为-ch3的对称变形振动吸收峰;1281.36cm-1和1243.41cm-1为酯基的c-o-c不对称伸缩振动峰;1114.20cm-1处为c-o-c伸缩振动峰;1062.22cm-1处为c-n伸缩振动峰;964.36cm-1处为=c-h的变形振动峰,843.33cm-1处为c-c的骨架振动峰。
(4)在分别在四口烧瓶中称取0.1mol四种不同分子量的甲氧基封端的二臂超支化聚氧乙烯醚大单体,充分溶解于水中,然后称取大单体总质量1%-5.6%的引发剂过硫酸铵,溶于水中得到溶液a,称取0.25-0.35mol丙烯酸和0.008-0.03mol甲基丙烯磺酸钠共同溶于水中得到混合溶液b。将温度升至60℃后开始向四口烧瓶中滴加a、b两种溶液,常压回流,机械搅拌,3h滴加完a、b两种液体,然后保温反应2h,冷却至室温,用30%naoh水溶液调节ph值至7左右,得到四种不同分子量的甲氧基封端的二臂超支化聚羧酸(1)、(2)、(3)、(4)。其中,甲氧基封端的二臂超支化聚羧酸(1)、(2)合成时甲基丙烯磺酸钠的加入量为0.03mol,甲氧基封端的二臂超支化聚羧酸(3)、(4)合成时甲基丙烯磺酸钠的加入量为0.008mol。图12为以分子量为400的mpeg为原料制备的二臂超支化聚羧酸的红外谱图。图中,3454.66cm-1处为聚羧酸分子内部缔合-oh的伸缩振动峰;2874.42cm-1处为饱和c-h键的伸缩振动峰;1726cm-1处为酯的羰基(c=o)特征峰;1566.51cm-1、1411.09cm-1处为羧酸盐中c-o的不对称伸缩振动峰及对称伸缩振动峰;1111.12cm-1处出现的信号峰为聚氧乙烯醚侧链中c-o-c的伸缩振动峰;1192.17cm-1处出现酯键的c-o-c的信号峰。
实施例2
所述的侧链超支化聚羧酸,以丙烯酸甲酯、马来酸酐为小单体,以巯基乙酸为链转移剂,以过硫酸铵为引发剂,通过水溶液自由基共聚反应,和甲氧基封端的四臂超支化聚氧乙烯醚大单体反应得到四臂超支化聚羧酸。
其制备方法如下:
(1)取0.1mol乙醇胺加入25ml甲醇,放入冰浴中,而后0.3mol丙烯酸甲酯加入25ml甲醇,放入恒压滴液漏斗中,冰浴氮气保护0.5h滴加完毕,而后继续冰浴反应0.5h,再油浴30℃黑暗条件下反应24h,30℃下减压蒸馏,得m0.5,m0.5的ftir谱图、1hnmr谱图和13cnmr谱图,见图4~6。图4中,3456.36cm-1是-oh的伸缩振动峰,2954.04cm-1是-ch3的不对称伸缩振动峰,2841.35cm-1是-ch2-对称伸缩振动峰,1736.30cm-1为酯基中c=o的特征峰,1438.22cm-1处为-ch2-的变形振动峰;1361.28cm-1处为-ch3的对称变形振动吸收峰,1257.08cm-1和1200.37cm-1为c-n的吸收峰,1037.12cm-1处是伯醇c-o的吸收峰,840.97cm-1处为c-c的骨架振动。图5中,3.69ppm处为甲氧基-o-ch3中氢的核磁共振峰,其余峰由于均为亚甲基-ch2-的质子峰,3.59ppm处是-oh邻位的亚甲基质子峰,2.98ppm处是-oh的质子峰,2.80ppm处是叔胺-n-邻位往羰基方向的亚甲基质子峰,2.60ppm处则是叔胺-n-邻位往羟基方向的亚甲基质子峰,2.47ppm处则是羰基c=o邻位往叔胺-n-方向的亚甲基质子峰。图6中,172.77ppm处为c=o碳的特征峰,58.88ppm处为-oh邻位碳原子特征峰,55.77ppm处为叔胺-n-邻位往羟基方向的碳原子特征峰,51.41ppm处为-o-ch3碳原子特征峰,49.0ppm处为叔胺-n-邻位往羰基方向的碳原子特征峰,33.37ppm处为羰基邻位往叔胺-n-方向的碳原子特征峰。
(2)取0.025mol的m0.5加入25ml甲醇,在冰浴条件下滴入0.6mol乙二胺和25ml甲醇的混合溶液中,氮气保护0.5h内滴加完毕,继续冰浴0.5h,后在30℃条件下氮气保护黑暗反应48h,55℃下减压蒸馏,得到四臂树状小分子m1;m1的ftir谱图、1hnmr谱图、13cnmr谱图,见图7~9。图7中,3395.15cm-1处为-oh的伸缩振动峰;2950.40cm-1处为-ch2-的不对称伸缩振动峰;1641.25cm-1处为酰胺中c=o的伸缩振动峰;1564.72cm-1处为n-h变形振动峰;1477.13cm-1处为-ch2-的变形振动峰;1326.31cm-1处为-oh的面内变形振动;1055.44cm-1处为c-n的伸缩振动峰。图8中,7.34ppm处-conh-的质子峰,3.50ppm处羟基邻位的-ch2-质子峰,3.13ppm处酰胺结构邻位的-ch2-质子峰,2.69ppm处胺基邻位-ch2-质子峰,2.60ppm处叔胺-n-邻位往酰胺方向的-ch2-质子峰,2.41ppm处叔胺-n-邻位往羟基方向的-ch2-质子峰,2.24ppm处酰胺结构邻位往叔胺-n-方向的-ch2-特征峰,以及2.08ppm处胺基-nh2和羟基-oh的质子峰。图9中,172.50ppm峰对应-conh-碳原子,58.59ppm峰对应-oh邻位碳原子,56.25ppm为叔胺-n-邻位羟基方向的碳原子特征峰,50.10ppm对应叔胺-n-邻位酰胺结构方向的碳原子,41.44ppm为酰胺结构-conh-邻位往胺基方向的碳原子峰,40.98ppm为胺基-nh2邻位往酰胺结构方向的碳原子峰,33.70ppm为酰胺-conh-邻位往叔胺-n-方向的碳原子峰。
(3)分别于两个三口烧瓶中取400(n=8)、600(n=13)两种分子量的mpeg0.1mol,0.2mol碳酸钾,放入100ml二氯甲烷中,回流条件下滴加0.5mol氯化亚砜和30ml二氯甲烷的混合液,45℃条件反应至完全,结束后用少量二氯甲烷冲洗回流冷凝管,并用砂芯漏斗过滤除盐,再减压蒸馏,得到两种不同分子量的cl-mpeg。
(4)分别取上述两种不同分子量的cl-mpeg0.1mol,放入100ml乙腈中,加入0.2mol碳酸钾和质量为总反应物质的质量1%的碘化钾和0.02molm1,85℃,黑暗条件下氮气保护回流24h,而后砂芯漏斗过滤,减压蒸馏,得到两种四臂接枝产物。
(5)将两种四臂接枝产物分别加入80ml四氢呋喃中,并再加入0.2mol碳酸钾。将丙烯酰氯0.06mol溶于20ml四氢呋喃中,置于恒压滴液漏斗中,冰浴滴加0.5h,而后黑暗氮气保护反应24h,过滤后减压蒸馏,即得两种甲氧基封端的四臂超支化聚氧乙烯醚大单体,以分子量为400的mpeg为原料得到的四臂超支化聚氧乙烯醚大单体的ftir谱图见图11。图11中,3455.81cm-1处为n-h伸缩振动峰;2877.04cm-1处为-ch2-的对称伸缩振动峰;1721.04cm-1处为酯的羰基(c=o)特征峰;1646.01cm-1处为c=c的伸缩振动峰;1456.87cm-1处为-ch2-的变形振动峰;1352.80cm-1处为-ch3的对称变形振动吸收峰;1298.98cm-1和1257.51cm-1为酯基的c-o-c不对称伸缩振动峰;1105.95cm-1处为c-o-c伸缩振动峰;1062.22cm-1处为c-n伸缩振动峰;952.46cm-1处为=c-h的变形振动峰;846.29cm-1处为c-c骨架振动峰。
(6)在两个四口烧瓶中分别称取0.1mol两种甲氧基封端的四臂超支化聚氧乙烯醚大单体,充分溶解于水中,然后称取大单体总质量1%-5.6%的引发剂过硫酸铵,溶水中得到溶液a,称取0.20mol丙烯酸甲酯和0.1mol马来酸酐和0.03mol链转移剂巯基乙酸共同溶于水中得到混合溶液b。将温度升至60℃后开始向四口烧瓶中滴加a、b两种溶液,常压回流,机械搅拌,3h滴加完a、b两种液体,然后保温反应2h,冷却至室温,用30%naoh水溶液调节ph值至7左右,得到两种不同分子量的甲氧基封端的四臂超支化聚羧酸(1)、(2)。图13为以分子量为400的mpeg为原料制备的四臂超支化聚羧酸的红外谱图。图中34555.43cm-1处为聚羧酸分子内部缔合-oh的伸缩振动峰;2874.55cm-1处为饱和c-h键的伸缩振动峰;1726cm-1处为酯的羰基(c=o)特征峰;1644.08cm-1处出现酰胺ⅰ带吸收峰;1561.54cm-1、1413.00cm-1处出现羧酸盐中c-o的不对称伸缩振动峰及对称伸缩振动峰;1110.89cm-1处的信号峰为聚氧乙烯醚侧链中c-o-c的伸缩振动峰。
对比例
本对比例选用传统直链醚型大单体甲基烯丙基聚氧乙烯醚(tpge),合成梳型聚羧酸。在四口烧瓶中称取400(n=7)、600(n=11)、1000(n=20)、2000(n=43)四种不同分子量的0.1mol醚型大单体tpeg,
tpeg的通式:
充分溶解于水中,然后称取大单体总质量1%-5.6%的引发剂过硫酸铵,溶于水中得到溶液a,称取0.25-0.35mol丙烯酸和0.008-0.03mol甲基丙烯磺酸钠共同溶于水中得到混合溶液b。将温度升至60℃后开始向四口烧瓶中滴加a、b两种溶液,常压回流,机械搅拌,3h滴加完a、b两种液体,然后保温反应2h,冷却至室温,用30%naoh水溶液调节ph值至7左右,得到四种不同分子量的传统梳型聚羧酸(1)、(2)、(3)、(4)。
其中传统梳型聚羧酸(1):n=7;传统梳型聚羧酸(2):n=11;传统梳型聚羧酸(3):n=20;传统梳型聚羧酸(4):n=43;x,y=20~200。
应用实施例
为了评价本发明所制备的侧链超支化聚羧酸的表面物化性能,对上述实施例1、实施例2及对比例中涉及的10种聚羧酸的表面张力、表观黏度及起泡性能进行了测试,测试结果见表1。
表1
从上表1可看出,本发明制备的侧链超支化聚羧酸在大单体聚合度相同的情况下,可一定程度上降低溶液的表面张力和表观黏度,有效提高其起泡性能,侧链四臂超支化聚羧酸降低幅度较二臂的程度更大。
以上所述实施例仅是为充分说明本发明而所举的较佳的实施例,本发明的保护范围不限于此。本技术领域的技术人员在本发明基础上所作的等同替代或变换,均在本发明的保护范围之内。本发明的保护范围以权利要求书为准。
- 还没有人留言评论。精彩留言会获得点赞!