Cmd1酶催化的5‑甲基胞嘧啶核酸的新修饰及其应用的制作方法
本发明属于表观遗传学领域,更具体地,本发明涉及cmd1酶催化的5-甲基胞嘧啶核酸的新修饰及其应用。
背景技术:
作为一种在动植物中广泛分布的核酸修饰,5-甲基胞嘧啶(5mc)与表观遗传调控等多种生物学过程息息相关。在哺乳动物中,5mc可以被tet双加氧酶氧化生成5-羟甲基胞嘧啶(5hmc),5-醛基胞嘧啶(5fc)和5-羧基胞嘧啶(5cac)。而5fc与5cac可以被tdg糖苷酶特异性识别并切除。在拟南芥中,5mc可以直接被特异的糖苷酶切除形成ap位点。这些5mc的加工过程都在一定程度上参与或促进了核酸去甲基化。在包括阿米巴原虫,灰盖鬼伞菌等生物中也同样存在5mc及类似的衍生化修饰,但是否存在其它不同的5mc衍生修饰还需要进一步研究。
利用进化上保守的tet-jbp结构域为模版,人们已经在多种物种中找到tet双加氧酶的同源蛋白。本发明人利用phytozome数据库在莱茵衣藻中鉴定出了8个tet的同源蛋白(crtet1-8,图1)。莱茵衣藻属于绿藻门,在进化上比较原始,被认为是许多植物的祖先。本发明人发现这8个tet同源蛋白都拥有与哺乳动物和阿米巴原虫的tet蛋白相一致的hxd模体,可能负责与fe2+的结合。虽然α-酮戊二酸(2-og)作为许多双加氧酶家族的必需辅因子参与酶活反应,本发明人并没有在这些蛋白中找到2-og的结合位点,暗示这些蛋白可能具有与众不同的特性。
因此,本领域有必要研究这些蛋白的酶活特性,找到它们的新功能,开拓它们的新应用。
技术实现要素:
本发明的目的在于提供cmd1酶催化的5-甲基胞嘧啶核酸的新修饰及其应用。
在本发明的第一方面,提供一种在甲基化核酸的5-甲基胞嘧啶(5mc)的甲基基团上加上甘油基团的方法,包括:cmd1酶处理甲基化核酸,从而在甲基化核酸的5-甲基胞嘧啶(5mc)的甲基基团上加上甘油基团。
在一个优选例中,以含有甘油基团的化合物作为甘油基的供体。
在另一优选例中,所述的含有甘油基团的化合物是维生素c或其类似物。
在另一优选例中,所述的维生素c类似物包括:脱氢抗坏血酸。
在另一优选例中,在以cmd1酶处理甲基化核酸的反应体系中,存在亚铁离子(fe2+)作为辅因子。
在另一优选例中,所述的在甲基化核酸的5-甲基胞嘧啶(5mc)的甲基基团上加上甘油基团的方法是非诊断性和治疗性的方法。
在另一优选例中,所述的在甲基化核酸的5-甲基胞嘧啶(5mc)的甲基基团上加上甘油基团的方法是体外方法。
在另一优选例中,cmd1酶处理甲基化核酸后,形成选自以下的产物(该产物存在于核酸链上)。
在另一优选例中,所述的cmd1酶是来源于藻类的cmd1酶;较佳地,所述的藻类是衣藻或团藻。
在另一优选例中,所述的cmd1酶包括:(a)如seqidno:1氨基酸序列的多肽;(b)将seqidno:1氨基酸序列经过一个或多个(如1-30个;较佳地1-20个;更佳地1-10个;如5个,3个)氨基酸残基的取代、缺失或添加而形成的,且具有(a)多肽功能的由(a)衍生的多肽;或(c)与(a)限定的多肽序列有60%以上(较佳地70%以上,如80%,85%,90%,95%,98%,99%或更高)同源性且具有(a)多肽功能的由(a)衍生的多肽;
并且,(b)~(c)限定的序列中,基于seqidno:1氨基酸序列,第330是a、第345位是h、第347是d、第350位是d。
在本发明的另一方面,提供一种cmd1酶或其上调剂的用途,用于在甲基化核酸的5-甲基胞嘧啶(5mc)的甲基基团上加上一个甘油基团;或用于制备在甲基化核酸的5-甲基胞嘧啶(5mc)的甲基基团上加上一个甘油基团的制剂。较佳地,所述的甘油基团来自含有甘油基团的化合物,例如维生素c或其类似物。
在一个优选例中,所述的cmd1酶的上调剂是可重组表达cmd1酶的表达载体。例如,pet28a载体,其中包含cmd1酶的基因表达盒。
在另一优选例中,所述的甘油基团通过碳碳单键连接到5-甲基胞嘧啶的甲基基团的碳原子上。
在本发明的另一方面,提供一种cmd1酶下调剂的用途,用于降低修饰的甲基化核酸水平,该修饰的甲基化核酸是5-甲基胞嘧啶的甲基基团上携带甘油基团的甲基化核酸。
在一个优选例中,所述的cmd1酶下调剂是降低cmd1基因表达或活性的物质;较佳地是特异性干扰cmd1基因表达的干扰分子,或是特异性抑制cmd1酶活性的抗体;更佳地,所述的干扰分子是cmd1基因或其转录本为抑制或沉默靶标的microrna或其前体、dsrna、反义核酸、小干扰rna,或能表达或形成所述microrna或其前体、dsrna、反义核酸、小干扰rna的构建物。
在另一优选例中,所述的cmd1酶下调剂是包括seqidno:10序列(cmd1amirna前体)的表达载体。
在另一优选例中,所述的cmd1酶或其上调剂或下调剂的用途是非诊断性和治疗性的用途。
在本发明的另一方面,提供一种cmd1酶下调剂,其是包括seqidno:10序列(cmd1amirna前体)的表达载体。
在本发明的另一方面,提供含有甘油基团的化合物的用途,用于为cmd1酶在甲基化核酸的5-甲基胞嘧啶的甲基基团上加上甘油基团过程中,作为甘油基团供体。例如,所述的含有甘油基团的化合物为维生素c或其类似物。
在一个优选例中,所述的含有甘油基团的化合物的用途是非诊断性和治疗性的用途。
在本发明的另一方面,提供一种鉴定甲基化核酸的方法,所述方法包括:
(1)以cmd1酶处理待测核酸(如基因组dna),从而使其中的甲基化核酸的5-甲基胞嘧啶(5mc)的甲基基团上加上甘油基团;
(2)鉴定反应产物中发生甘油基团修饰的产物,从而确定甲基化核酸的存在情况或存在量。
在一个优选例中,步骤(1)中,存在含有甘油基团的化合物作为甘油基团的供体,并且存在亚铁离子(fe2+)作为辅因子;较佳地,所述的含有甘油基团的化合物是维生素c或其类似物。
在另一优选例中,所述的甘油基团上进行了标记;较佳地,所述的标记是13c同位素标记。
在另一优选例中,所述的鉴定甲基化核酸的方法是非诊断性和治疗性的方法。
在本发明的另一方面,提供一种检测甲基化核酸上基因修饰的单碱基检测方法,所述方法包括:
(a)对于待测核酸,采用tet双加氧酶处理,从而5-甲基胞嘧啶(5mc),5-羟甲基胞嘧啶(5hmc)以及5-醛基胞嘧啶(5fc)均被氧化成5-羧基胞嘧啶(5cac),而具有甘油基团修饰的5-甲基胞嘧啶(5mc)未被氧化;和
(b)采用重亚硫酸氢盐测序法测定,将待测核酸中的被脱氨转化的5-甲基胞嘧啶等与具有甘油基团修饰而未被转化的的5-甲基胞嘧啶区分开。
在一个优选例中,所述的检测甲基化核酸上基因修饰的单碱基检测方法是非诊断性和治疗性的方法。
在本发明的另一方面,提供一种单碱基检测方法,所述方法包括:
(s1)对于待测核酸,利用第三代或更新的核酸测序方法进行测序,从而区分出待测核酸中的胞嘧啶与5-羟甲基胞嘧啶(5hmc),5-醛基胞嘧啶(5fc)与5-羧基胞嘧啶(5cac);较佳地,所述的第三代或更新的核酸测序方法包括并不限于smrtsequencing或nanoporesequencing;
(s2)以cmd1处理待测核酸,在5-甲基胞嘧啶(5mc)的甲基基团上加上甘油基团,从而区分出待测核酸中的5-甲基胞嘧啶(5mc)。
在本发明的另一方面,提供一种区分待测核酸中5-甲基胞嘧啶(5mc)与5-羟甲基胞嘧啶(5hmc)的方法,所述待测核酸中不存在甘油基团修饰的5-甲基胞嘧啶(5mc),所述方法包括:
(i)以cmd1酶处理所述待测核酸,从而5-甲基胞嘧啶(5mc)被转变为甘油基团修饰的5-甲基胞嘧啶(5mc),而5-羟甲基胞嘧啶(5hmc)不能被甘油基团修饰;
(ii)采用tet双加氧酶处理步骤(i)的产物,5-羟甲基胞嘧啶(5hmc)被氧化成5-羧基胞嘧啶(5cac),而甘油基团修饰的5-甲基胞嘧啶(5mc)未被氧化,从而区分待测核酸中5-甲基胞嘧啶(5mc)与5-羟甲基胞嘧啶(5hmc);较佳地通过bisulfite-seq方法区分。
在一个优选例中,所述的区分待测核酸中5-甲基胞嘧啶与5-羟甲基胞嘧啶的方法是非诊断性和治疗性的方法。
在本发明的另一方面,提供一种测定待测样品中是否存在含有甘油基团的化合物的方法,所述方法包括:在待测样品中加入cmd1酶与含5mc的核酸,测定其中甲基化核酸的5-甲基胞嘧啶(5mc)的甲基基团上是否加上甘油基团;若是,则表明待测样品中存在含有甘油基团的化合物。例如,所述的含有甘油基团的化合物为维生素c或其类似物。
在本发明的另一方面,提供一种靶向性调控基因表达的方法,所述方法包括:以cmd1酶处理靶基因,从而在甲基化核酸的5-甲基胞嘧啶的甲基基团上加上甘油基团,获得5-甲基胞嘧啶被甘油基修饰的基因,该基因的表达与cmd1酶处理前相比,表达发生变化。所述的靶向性调控基因表达的方法可用于细胞的转录调控;较佳地,特异性抑制原癌基因或其它致病性基因等;或可用于控制基因表达而调控细胞分化等。
在本发明的另一方面,提供一种通过改变细胞基因组或体外核酸的修饰形式而影响蛋白结合或酶活的方法,其特征在于,所述方法包括:cmd1酶处理细胞内源基因组或体外核酸,从而在其中甲基化核酸的5-甲基胞嘧啶的甲基基团上加上甘油基团,获得甘油基修饰的5-甲基胞嘧啶,从而影响细胞基因组或体外核酸与蛋白的结合,或影响酶作用于细胞基因组或体外核酸的功能。例如,所述的酶是限制性内切酶;更具体地例如mspi。
在本发明的另一方面,提供一种用于在甲基化核酸的5-甲基胞嘧啶的甲基基团上加上甘油基团的试剂盒,该试剂盒中包括:cmd1酶或其上调剂;含有甘油基团的化合物;较佳地,所述的含有甘油基团的化合物是维生素c或其类似物,或能产生vc的物质;和亚铁离子或能形成亚铁离子的物质。
本发明的其它方面由于本文的公开内容,对本领域的技术人员而言是显而易见的。
附图说明
图1、莱茵衣藻中tet同源蛋白与阿米巴原虫tet1的氨基酸序列比对图谱。八个衣藻tet同源蛋白是利用tet-jbp结构域在衣藻基因组中比对得到,这些蛋白和阿米巴原虫以及哺乳动物中的tet蛋白一样,都有保守的hxd基序。序列上方的标记表示该位置的氨基酸残基在ngtet1结构和生化分析中被验证过所具有的特定功能:“m”表示金属离子(亚铁离子)结合位点,“c”表示与5mc相互作用位点,“a”表示活性中心位点,“α”表示α-酮戊二酸结合位点。α-酮戊二酸结合位点在衣藻tet同源蛋白crtet1(cmd1)中不保守。
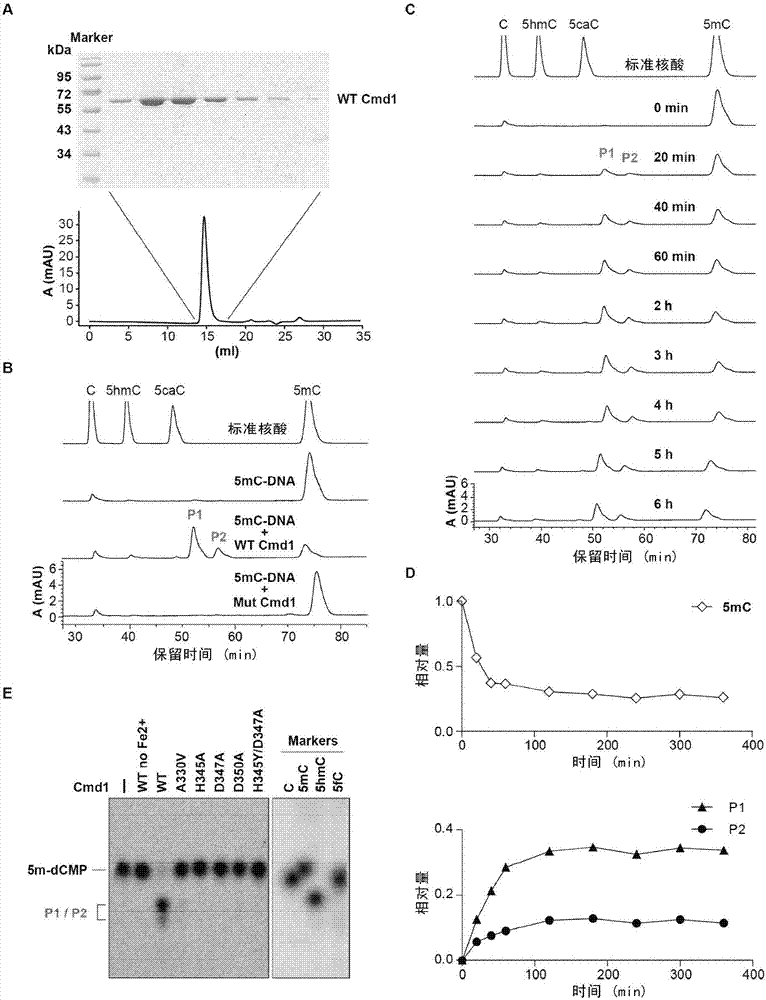
图2、cmd1催化5mc形成新的dna修饰。
a:从大肠杆菌中纯化到的不含标签cmd1的考马斯亮蓝染色图。图片展示在凝胶过滤层析中不同收集管(14-17min,1ml/min)中的cmd1蛋白。
b:hplc分析5mc-dna与cmd1蛋白孵育之后的dna成分。p1/p2表示2个新检测到的与标准品不同的峰。mutcmd1是cmd1的酶活突变体,其含有两个点突变(h345y/d347a)。
c:hplc结果显示,在5mc-dna底物与cmd1孵育的前两个小时,产物p1和p2不断累积并逐渐达到平台期。
d:cmd1酶活反应的时间曲线,5mc、p1以及p2的相对含量由hplc峰面积(图2c)计算得出。
e:tlc检测5mc-dna经cmd1修饰后的核苷成分。5mc的甲基用14c标记后与野生型或突变型cmd1反应。p1/p2表示tlc板上检测到的新的修饰点。
图3、当所预测的功能性氨基酸残基发生突变后,cmd1的大部分或全部酶活丧失。
a:大肠杆菌中纯化的cmd1及其突变体的考马斯亮蓝染色图。根据序列比对结果分析,提示h345和d347是与亚铁离子结合的位点,a330是cmd1的酶活中心位点,d350与5mc相互作用。
b:cmd1突变体几乎丧失催化5mc到p1或p2的酶活性。
c:含有5mc,而非含c或5hmc的dna能够作为cmd1催化反应的底物。通过pcr扩增得到含有c、5hmc或5mc的dna,经过cmd1孵育后,用hplc检测其核苷组成。p1和p2核苷在含5mc的dna与野生型cmd1(wtcmd1)孵育时被检测到,关键区域突变的cmd1则丧失活性。mutcmd1是cmd1的酶活突变体,其蛋白序列含有两个点突变(h345y/d347a)。
图4、p1/p2的分子式与结构式的解析。
a:质谱检测p1/p2核苷的相对分子质量。p1/p2核苷是由cmd1体外催化5mc-dna经酶法水解,再通过hplc分离得到的。216.0976峰刚好对应于核苷p1/p2(332.1448,理论上)在中性丢失脱氧核糖(116)之后剩余的碱基部分的分子量。
b:根据二维核磁共振以及密度泛函理论所确定的p1/p2的结构式。p1与p2是立体异构体,仅在c8位置存在手性差异。
图5、氘代示踪实验显示cmd1反应后5mc甲基上氢原子的去向。
a:质谱检测dna底物中甲基被氘(d)特异性标记的5mc(d3标记的5mc)。该dna底物在体外用m.sssi和d3标记的s-(5’-腺苷)-l-甲硫氨酸(sam)反应制备。甲基上的氘代5mc核苷与未被标记的相比,相对分子质量增加3个单位。
b:质谱检测由d3标记5mc产生的p1。相对于来源未标记的5mc的p1核苷,氘代5mc产生的p1相对分子质量增加了2个单位。
c:根据串联质谱测定到的各种离子的质荷比,推测p1/p2碱基及其碎片的结构。p1和p2产生质量完全相同的碰撞诱导解离反应(cid)碎片,这暗示着它们很可能是同分异构体。图片展示p1/p2丰度最高的一些cid碎片,根据测定的相对分子质量推算得到对应的分子式与结构式。由于所有来源于d3标记5mc的p1或p2的碎片离子比来源于未标记5mc的p1或p2的碎片离子都增加了2个单位,所以新修饰极有可能发生在5mc的甲基上;通过cid结果来看,与嘧啶环连接的亚甲基在cid过程中并没有发生改变。在cid中,p1/p2出现质量连续三次下降18.01的碎片,,暗示在p1和p2结构上存在三个羟基,在质谱分析过程中发生了相当于三次脱水(mw18.0100)的反应。
图6:维生素c(vc)直接参与5mc的修饰反应。
a:p修饰中增加的90dalton部分不是来源于cmd1或与cmd1共纯化的化合物。该实验所用cmd1蛋白是从生长在仅含12c或13c标记的葡萄糖作为唯一碳源的m9基本培养基中的大肠杆菌中纯化得到。13c-cmd1产生的p1与12c-cmd1产生的p1相比,相对分子质量并没有增加,这也暗示产生p1所需要的反应物并非来源于cmd1及共纯化的化合物,而是来源于酶活反应中的某个成分。
b:cmd1酶活对各个因子的依赖性检测。图中标示出cmd1在缺少不同的小分子条件下的酶活反应后用hplc检测的结果。vc在这个反应中是必需的。
c:cmd1的酶活不依赖于2-og。利用2-og的竞争性抑制剂n-oxalylglycine(n-og),并不影响cmd1的酶活。
d:2-og和fe2+是htet2酶活所必需的,而维生素c是非必需的。酶活反应条件差异见标注,之后用hplc检测dna中的核苷组成。n-og能够抑制htet2活性。
e:hplc检测氧化型vc,脱氢抗坏血酸(dha)对cmd1的酶活影响。dha只有在利用dtt将其还原成vc后,才可以支持cmd1的酶活。
f:hplc检测vc的多种类似物对cmd1的酶活影响。
g:hplc检测经热失活处理的vc对cmd1的酶活影响情况。
h:hplc检测fe2+以及其它二价金属离子对cmd1的酶活影响。
图7、vc是cmd1催化5mc产生新修饰过程中的甘油基团供体。
a:vc直接参与5mc的修饰。质谱结果显示13c6-vc替换不含同位素标记的vc产生的p1在分子量上会增加了3daltons。
b:质谱结果显示[1-13c]-vc和[3-13c]-vc没有造成p1分子量的改变。
c:cmd1反应中,vc的4到6号碳原子部分会被转移到5mc的甲基上。当用[5-13c]-vc或[1,6-13c2]-vc替换未标记的vc时,产生的p1在分子量上会增加。图谱红色标记显示了在不同标记的vc产生的p1中含有13c原子的最小分子碎片。
图8:vc参与cmd1酶活反应的机理与反应产物检测。
a:hplc检测在无氧的手套箱中进行的cmd1酶活反应结果。实验显示,氧气对cmd1的酶活是不可或缺的。
b:质谱检测利用18o标记的氧气或水进行cmd1酶活反应产生的p1的分子量。
c:cmd1酶活反应机理预测。fe(ii)首先与cmd1蛋白上的his-x-asp-xn-his保守结构域配位,另三个配位键则被水分子占据(i)。vc水解后,利用c-1上的羧基与c-2上的羰基与fe(ii)螯合,替换掉2个水分子(ii)。当5mc进入cmd1的酶活中心附近后,fe(ii)上另一个水分子脱离(iii)。氧气与fe(ii)中心的结合产生了fe(iii)-superoxo超氧化物中间体(iv)。该中间体的远端氧亲核进攻vc水解产物c-2位上的羰基,产生了fe(iv)-peroxo过氧化物基团(v)。之后起始的氧化脱羧又形成fe(iv)-oxo基与羧酸的配位中间体(vi)。此活性基团从5mc中夺取一个氢原子,形成fe(iii)-oh中间体与5mc自由基(vii)。配位中间体上的c-2羟基螯合到fe(iii)中心,产生的水分子随即脱离(viii)。5mc自由基进攻该配位中间体,造成c2-c3碳碳键均裂,形成了p核苷以及与fe(ii)结合的乙醛酸(ix)。乙醛酸随即解离,fe(ii)回复到初始状态并完成整个反应循环。
d:气相色谱实验检测13c-vc所释放出的co2。
e:质谱检测cmd1反应产生的乙酸酸。乙醛酸由于分子量低,极性大,因此经过dnp衍生后再进行质谱检测。
图9、在cc125衣藻品系细胞中也存在p修饰碱基,其含量变化依赖于cmd1的表达水平。
a/b:三重四级杆质谱检测基因组dna中的p修饰以及5mc的含量。p表示p1与p2的总和,因为在本质谱实验的条件下无法有效区分p1与p2。
c/d:在cmd1knockdown藻株中检测p修饰碱基以及5mc的含量。**,p<0.01;***,p<0.001。结果提示cmd1酶量减少,产生的p修饰减少,5mc含量会上升。
e/f:用5-氮杂-2’-脱氧胞苷(5-aza-2’-deoxycytidine,5-aza,一种胞嘧啶甲基转移酶抑制剂)处理后,衣藻细胞中的5mc和vc衍生出的p修饰水平明显下降。*,p<0.05;***,p<0.001。在5-aza处理的不同细胞株中,cmd1的表达水平并没有发生改变。
图10、在cc125衣藻品系细胞中对cmd1进行knockdown。
a:cmd1amirna重组质粒的构建以及cmd1高knockdown效率细胞株的筛选策略。
b:三株knockdown细胞株(ami-cmd1-1-3)和一株对照细胞株(ami-ctrl)的相对荧光素酶活性检测。
c:三株knockdown细胞株(ami-cmd1-1-3)和一株对照细胞株(ami-ctrl)中cmd1基因的相对表达水平。cmd1基因的相对表达水平通过q-pcr获得,以cblp基因的表达水平作为内参。图片中展示的是来自三组独立重复实验数据的平均标准误。
图11、cmd1催化反应方程式。在cmd1催化反应中,维生素c作为辅助底物提供一个甘油基团,该基团会被转移到dna中5mc的甲基上。在体外,甘油基团与5mc相连的碳原子(c8)在成键时会形成两种手性的异构体,p1和p2。乙醛酸与co2是vc反应产生的2种副产物。
图12、cmd1产生的5mc新修饰(p修饰)影响基因表达与胞嘧啶修饰的单碱基水平检测。
a:报告基因表达检测显示p修饰抑制基因表达。在hek293t细胞中,启动子上的p修饰显著影响了firefly报告基因的表达,共转tet2与tdg也无法激活此报告基因。
b:p修饰在基于tet氧化以及bisulfite-seq(tet-oxi-bs-seq)的、针对胞嘧啶修饰状态的单碱基检测中的作用示意图。重亚硫酸氢盐处理耦联pcr扩增(bisulfitepcr,bs-pcr)后,tet氧化以及bisulfite处理后的dna测序,只有p碱基一种以“c”的形式读出。
c:cobra(combinedbisulfiterestrictionanalysis重亚硫酸氢盐处理耦联pcr扩增并限制性内切酶分析)数据显示,dna中的5mc因为体外发生甘油化修饰而免受脱氨转化,在pcr扩增序列中保持了c的形式,从而能被taii限制性核酸内切酶(识别acgt序列)切开。
d:bisulfite-seq检测体外酶活产生的p修饰碱基。图中每一个圈表示一个cpg位点。向下的小箭头标明taii切割位点。control(ctrl)5mc-dna在tet-oxi-bs-seq中,有6.1%以c(橙色)的形式读出,说明tet氧化或之后的脱氨转化不完整。但5mc-dna被cmd1处理后,极大部分5mc(约80%)都被保护了。
图13、cmd1介导的p修饰影响了限制性内切酶mspi(识别ccgg序列)对5mc-dna的切割活性。
具体实施方式
5-甲基胞嘧啶(5mc)是在核酸胞嘧啶5位碳原子上发生甲基化形成的一种表观遗传修饰,在动植物生长发育与疾病发生发展中起重要作用。本发明人首次发现一种5mc修饰酶(cmd1),可以在甲基化核酸的5mc甲基碳上通过碳碳单键连接一个甘油基团。在此基础上完成了本发明。
如本文所用,除非另外说明,所述的“甲基化核酸”是指一种核酸,其核酸链的胞嘧啶上,存在5位碳原子(c)上的甲基化修饰。核酸甲基化能引起染色质结构、核酸构象、核酸稳定性及核酸与蛋白质相互作用方式的改变,基因转录的改变(如使得基因沉默)从而控制基因表达。
如本文所用,除非另外说明,所述的“cmd1酶”即衣藻tet1。较佳地,所述的cmd1酶或其同源蛋白存在于藻类中,包括但不限于衣藻、团藻等。
如本文所用,所述的核酸包括:脱氧核糖核酸(dna)和核糖核酸(rna)。
本发明首次揭示了一种在甲基化核酸的5-甲基胞嘧啶(5mc)的甲基基团上加上甘油基团的方法,包括:cmd1酶处理甲基化核酸,从而在甲基化核酸的5-甲基胞嘧啶(5mc)的甲基基团上加上甘油基团。所述的方法可以是体内方法或体外方法。
cmd1的酶活需要fe2+作为辅因子,而维生素c(vc)作为一种必需的辅因子直接参与cmd1催化的反应,并作为甘油基的供体来源。
cmd1酶处理甲基化核酸后,形成选自以下的产物:
本发明人在体外水平和体内水平均检测到了这种新修饰的存在。通过降低cmd1基因的表达水平,这种修饰的含量也同时降低。
在本发明中,所述的cmd1酶可以是天然存在的,比如其可被分离或纯化自哺乳动物。此外,所述的cmd1酶也可以是人工制备的,比如可以根据常规的基因工程重组技术来生产重组cmd1酶。较佳地,可采用重组的cmd1酶。
来源于其它物种的cmd1酶均包含于本发明中,特别是那些与cmd1酶具有同源性的酶。较佳地,所述的其它物种中的相应cmd1酶序列与本发明中seqidno:1序列相同性高于60%的,更佳地是高于70%,更佳地是高于80%,更佳地是高于85%,更佳地是高于88%,更佳地是高于90%,更佳地是高于95%,更佳地是高于98%。
任何适合的cmd1酶均可用于本发明。所述的cmd1酶包括全长的cmd1酶或其生物活性片段(或称为活性片段)。
经过一个或多个氨基酸残基的取代、缺失或添加而形成的cmd1酶的衍生物或其生物活性片段也包括在本发明中,只要其保留有野生型cmd1酶的功能。cmd1酶或其生物活性片段包括一部分保守氨基酸的替代序列,所述经氨基酸替换的序列并不影响其活性或保留了其部分的活性。适当替换氨基酸是本领域公知的技术,所述技术可以很容易地被实施,并且确保不改变所得分子的生物活性。这些技术使本领域人员认识到,一般来说,在一种多肽的非必要区域改变单个氨基酸基本上不会改变生物活性。见watson等molecularbiologyofthegene,第四版,1987,thebenjamin/cummingspub.co.p224。
任何一种cmd1酶的生物活性片段都可以应用到本发明中。在这里,cmd1酶的生物活性片段的含义是指作为一种多肽,其仍然能保持全长的cmd1酶的全部或部分功能。通常情况下,所述的生物活性片段至少保持50%的全长cmd1酶的活性。在更优选的条件下,所述活性片段能够保持全长cmd1酶的60%、70%、80%、90%、95%、99%、或100%的活性。
一旦分离获得了所述蛋白的序列,就可以用重组法来大批量地获得该蛋白。这通常是将其编码基因克隆入载体,再转入细胞,然后通过常规方法从增殖后的宿主细胞中分离得到。此外,对于较短的蛋白而言,也可采用人工合成(如通过多肽合成仪合成)的方法来合成有关序列,人工合成的方法可简便且快速地得到所需要的蛋白。
本发明还包括了编码所述的cmd1酶的生物活性片段的分离的核酸,也可以是其互补链。编码cmd1酶的生物活性片段的dna序列,可以全序列人工合成,也可用pcr扩增的方法获得。在获得了编码所述的cmd1酶的生物活性片段的dna序列之后,将其连入合适的表达载体,再转入合适的宿主细胞。最后通过培养转化后的宿主细胞,通过分离纯化得到所要的蛋白。
本发明还包括了包含编码所述cmd1酶的生物活性片段的核酸分子的载体。所述的载体还可包含与所述核酸分子的序列操作性相连的表达调控序列,以便于蛋白的表达。所述的“操作性相连”或“可操作地连于”指这样一种状况,即线性dna序列的某些部分能够调节或控制同一线性dna序列其它部分的活性。例如,如果启动子控制序列的转录,那么它就是可操作地连于编码序列。
此外,含有编码所述cmd1酶的生物活性片段核酸序列的重组细胞也包括在本发明中。“宿主细胞”包括原核细胞和真核细胞。常用的原核宿主细胞包括大肠杆菌、枯草杆菌等;例如可为大肠杆菌细胞(e.coli),如大肠杆菌hms174(de3)、或bl21(de3)。常用的真核宿主细胞包括酵母细胞、昆虫细胞和哺乳动物细胞。
基于本发明人的新发现,本发明提供了cmd1酶或其生物活性片段的用途,包括但不限于:用于在甲基化核酸的5-甲基胞嘧啶的甲基基团上加上一个甘油基团;或用于制备在甲基化核酸的5-甲基胞嘧啶的甲基基团上加上一个甘油基团的制剂,等。
如本文所用,所述的cmd1酶的上调剂包括了稳定剂、激动剂、促进剂等。任何可提高cmd1酶的活性、维持cmd1酶的稳定性、促进cmd1酶的表达、延长cmd1酶有效作用时间、促进cmd1酶的基因转录和翻译以及的物质均可用于本发明,作为可用于调节(如促进)甲基化核酸的5-甲基胞嘧啶的甲基基团上的甘油基团修饰的有效物质。
如本文所用,所述的cmd1酶的下调剂包括了拮抗剂、抑制剂、阻滞剂、阻断剂等。任何可降低cmd1酶的活性、降低cmd1酶的稳定性、抑制cmd1酶的表达、减少cmd1酶有效作用时间或抑制cmd1酶的基因转录和翻译的物质均可用于本发明,作为可用于调节(如降低)甲基化核酸的5-甲基胞嘧啶的甲基基团上的甘油基团修饰的有效物质。
基于本发明的新发现以及结合现有常识,本领域技术人员可以制备或设计出cmd1酶的激动剂或下调剂,这些激动剂或下调剂也包括在本发明中。
作为本发明的优选方式,所述的cmd1酶的上调剂包括(但不限于):表达cmd1酶的质粒。
所述的cmd1酶的上调剂可通过影响cmd1酶或其编码基因的表达或活性而实现对甲基化核酸的5-甲基胞嘧啶的甲基基团上的甘油基团修饰的调控。
所述的cmd1酶的下调剂可以是核酸抑制物,蛋白抑制剂,抗体,配体,蛋白水解酶、蛋白结合分子、edta,其能够下调cmd1酶的表达或活性。作为本发明的优选方式,所述的cmd1酶的下调剂是基于cmd1酶的编码基因的序列设计的核酸抑制物,如小干扰分子。所述的干扰rna分子可通过采用适当的转染试剂被输送到细胞内,或还可采用本领域已知的多种技术被输送到细胞内。作为本发明的优选方式,所述的干扰分子是artificialmicrorna(amirna),一些优选的amirna被揭示于实施例中。根据已知的蛋白,筛选可以抑制其活性的抗体(单抗或多抗)也是本领域常规的技术。
所述的cmd1酶的下调剂通过影响cmd1酶的表达或活性而实现对核酸甲基化的调控。
作为一种实施方式,提供了一种cmd1酶下调剂的用途,用于降低修饰的甲基化核酸水平,该修饰的甲基化核酸是5-甲基胞嘧啶的甲基基团上携带甘油基团的甲基化核酸。
本发明揭示了cmd1介导全新的表观遗传修饰,同时也指出了5mc与vc在这种情况下新的命运决定因素。由于cmd1特异性地修饰甲基化的核酸,因而cmd1介导的这种修饰可以用来阻止或促进进一步的酶学或化学修饰,一方面可以用于基于改变基因功能的基因工程或基因治疗手段的开发,另一方面可以用于与体内核酸甲基化谱式检测相关的诊断技术的开发。
基于本发明的新发现,可以将本发明应用于鉴定甲基化核酸的存在情况或存在量,所述方法包括:(1)以cmd1酶处理待测核酸(如基因组dna),从而使其中的甲基化核酸的5-甲基胞嘧啶的甲基基团上加上甘油基团;(2)鉴定反应产物中发生甘油基团修饰的产物,从而确定甲基化核酸的存在情况或存在量。可以在甘油基团的供体上标记上可识别的标志物,以用于鉴别发生这种特殊修饰的5-甲基胞嘧啶。该方法中,反应体系中还包括维生素c作为甘油基团的供体,并且存在亚铁离子(fe2+)作为辅因子。与维生素c具有相同功能的维生素c的类似物也被包含在本发明中,只要其也具有甘油基可可用作供体。本领域技术人员了解一些公知的维生素c类似物,包括但不限于脱氢抗坏血酸等。
基于本发明的新发现,可以将本发明应用于检测甲基化核酸上基因修饰的单碱基检测方法。现在常用bisulfite-seq(重亚硫酸氢盐测序)的方法来检测核酸上5mc的存在。在传统的bisulfite-seq中,c,5fc与5cac发生脱氨形成u,在pcr扩增中被读出为t;而5mc与5hmc无法发生脱氨,因此经pcr扩增后仍被读出为c,从而与c等区分开来。但传统的bisulfite-seq无法区分5mc与5hmc,同样也无法区分c,5fc与5cac。并且由于p修饰经bisulfite-seq读出的结果与5mc一致,因此也无法与5mc进行区分。因此,本发明人提出了tet双加氧酶氧化耦联的bisulfite-seq方法,即首先用tet双加氧酶将5mc,5hmc以及5fc全部氧化成5cac,而p修饰无法被tet氧化,因此经bisulfite-seq就可以区分5mc与p修饰。
而同时,在核酸(如gdna)中不存在p修饰的情况下,利用这种方法也可以起到区分5mc与5hmc的作用。首先,用cmd1对核酸进行处理,将5mc全部转变为p修饰,而5hmc仍保持原样。再进一步利用tet双加氧酶氧化耦联bisulfite-seq的方法,从而将5mc与5hmc区分开来。
cmd1催化5mc与vc反应生成的新产物,很显然可被用在最新的核酸测序中检测5mc的含量。
本发明的技术方案结合第三代或更新的核酸测序技术,包括并不限于smrtsequencing以及nanoporesequencing等,可以直接区分5mc以及甘油基修饰的5mc,从而达到单碱基测定甘油基修饰的5mc的目的。目前,第三代测序技术暂时无法区分5mc与c,但可以区分c与5hmc,5fc与5cac;因此在第三代或更新的测序技术的基础上,以cmd1处理来特别鉴定5mc,从而可实现将c、5mc、5hmc、5fc、5cac同时加以区分。
此外,cmd1蛋白可用于检测vc的存在。因为vc在此反应中是必需的,不可替代的,因此可用cmd1蛋白与5mc-核酸来检测vc的存在,灵敏度和特异性都很高。
本发明的方法结合新一代核酸测序(第三代或更新的测序)技术,可使同时测定序列和修饰成为可能。
本发明的方法还为定点改变核酸片段或基因的结构提供了新的途径,还可在多种dnananotechnology中发挥作用。本发明的方法还可被应用于有机合成中碳链的延伸以及甘油基化合物的合成中。
基于本发明的新发现,还提供一种靶向性调控基因表达的方法,所述方法包括:基于cmd1介导的5mc新修饰可显著抑制基因表达,cmd1可与基因靶向性元件联用;较佳地,所述靶向性基因表达调节可通过crispr-cas9或talen结合cmd1酶而实现,可用于癌基因等的靶向性调控来实现治疗。此外,通过crispr-cas9或talen与cmd1联用,也可以用于降低靶基因的5mc修饰水平。
基于本发明的新发现,还提供一种改变细胞内源基因组上或体外dna上影响蛋白结合或酶活发挥的方法。由cmd1介导的甘油基修饰5mc改变了5mc原有的性质,从而影响其与蛋白的结合。例如,经cmd1修饰之后可用于改变限制酶内切酶如mspi等的酶活。
基于本发明的新发现,还提供了一种用于在甲基化核酸的5-甲基胞嘧啶的甲基基团上加上甘油基团的试剂盒(或称为药盒),其中包括:cmd1酶或其上调剂。更佳地,所述试剂盒中还包括:cmd1酶或其上调剂;维生素c;和亚铁离子或能形成亚铁离子的物质。所述的试剂盒将试剂集成于一个套组中,便于它们的商业应用。
作为本发明的优选方式,所述的试剂盒中还可包括其它一些试剂,例如定量或定性分析核酸甲基化情况的试剂等。更佳地,所述的试剂盒中还可包括使用说明书,以便于人们使用。
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件如j.萨姆布鲁克等编著,分子克隆实验指南,第三版,科学出版社,2002中所述的条件,或按照制造厂商所建议的条件。
材料与方法
1、cmd1重组蛋白的表达与纯化
本发明人将cmd1基因的编码序列克隆到改造过的pet28a表达载体(ppei-his-sumo,获自复旦大学徐彦辉实验室)上,并在大肠杆菌bl21(de3)中进行表达。
cmd1突变体的编码序列也被克隆到相同的表达载体。cmd1氨基酸序列如seqidno:1;以该序列为基准,cmd1突变体分别发生a330v突变,h345a突变,d347a突变,d350a突变,h345y和d347a(h345y/d347a)突变。并将重组表达载体转入大肠杆菌bl21(de3),进行诱导性表达。
培养大肠杆菌到od600达到0.8时,加入终浓度0.1mm的异丙基-β-d-硫代半乳糖苷(iptg),16℃诱导16h。将蛋白提取物中带有his-sumo标签的cmd1融合蛋白先与ni-nta纯化试剂(qiagen)结合,再与his-ulp1蛋白酶在4℃孵育过夜,以切除融合蛋白中的his-sumo标签。收集酶切液,并利用resource15q阴离子交换柱(gehealthcare)纯化,以缓冲液a(20mmtris-hcl,ph8.5)/缓冲液b(20mmtris-hcl,ph8.5,1mnacl)从100/0到50/50的线性梯度进行洗脱。洗脱液通过凝胶过滤层析进一步纯化,所使用的凝胶柱和缓冲液分别为superdex20010/300gl凝胶柱(gehealthcare)和缓冲液c(20mmhepesph7.0,100mmnacl)。凝胶过滤层析的洗脱液使用ultracel-10k超滤管(millipore)浓缩,使蛋白浓度达到10μg/μl。
2、cmd1体外酶活所需dna底物的制备
以莱茵衣藻(chlamydomonasreinhardtii)基因组dna为模板,以5m-dctp替代常规dctp,通过pcr扩增一段1.1kb的5mc-dna片段。为了测试cmd1对反应底物的特异性,5hm-dctp以及未修饰的dctp也用来通过pcr扩增获取相似的产物。pcr所使用的上下游引物分别为:
5’-biotin-aagggttggattgtaggtagtttagaaat-3’(seqidno:11),和
5’-tgagggtggtaaattag-3’(seqidno:12)。
3、cmd1体外酶活检测
在100μl反应体系中,0.5μg生物素标记的5mc-dna底物和4μgcmd1酶(酶与5mc的摩尔比约为1:2)在含有50mmhepes(ph7.0)、50mmnacl、1mml-抗坏血酸、1mmatp、1mmdtt和0.1mmfe(nh4)2(so4)2的体系中37℃反应1h。在13c示踪实验中,13c标记的l-抗坏血酸购自omicronbiochemicals。反应结束后,向反应体系中加入蛋白酶k以消化蛋白,再利用streptavidinsepharose纯化试剂(gehealthcare)对生物素标记的dna进行纯化,具体过程参照使用说明书。
4、高效液相层析色谱法(hplc)检测cmd1反应后dna底物的核苷组成
hplc检测方法包括:纯化后的生物素标记的dna底物用核酸酶p1(sigma)在含有0.2mmznso4和20mmnaac(ph5.3)体系中55℃消化至少1h,之后加入ciap(takara)及其缓冲液37℃反应1h来去除磷酸基。样品离心后取上清进行hplc分析,所使用的仪器型号为agilent1260,分析柱为welchaq-c18column(4.6x250mm,5μm)。检测核苷组成所使用的流动相为10mmkh2po4(ph3.85),流速为0.6ml/min,温度为15℃;回收p修饰核苷所使用的流动相为20mmnh4ac(ph5.21),流速1ml/min,温度为15℃。检测波长为280nm。
5、制备含甲基上标记14c或氘(d)的5mc-dna
在含有8μls-[甲基-14c]-l-甲硫氨酸(14c-sam,1.48-2.22gbq/mmol,perkinelmer)或s-[甲基-d3]-腺苷-l-甲硫氨酸(d3-sam,zzstandard)的100μl反应体系中,将5μg质粒dna和20unitsm.sssicpg甲基转移酶(zymoresearch)30℃孵育过夜。反应后被标记的dna使用qiaquicknucleotideremovalkit(qiagen)进行纯化和回收。
6、薄层层析法(tlc)检测5mc衍生物
将含有14c-5mc-dna底物与cmd1孵育,经蛋白酶k消化后,用酚氯仿和乙醇抽提纯化dna。将dna沉淀溶于8μl水中,用核酸酶p1进行裂解,取0.5μl裂解液点样于pei-cellulosetlc板(merck)上。带有样品的tlc板用异丙醇:hcl:h2o(70:15:15)进行展开,最后使用磷屏压片,并在fujifilmfluorescentimageanalyzerfla-3000上扫描成像。
7、液相色谱偶联质谱法(lc-ms)测定p修饰在衣藻基因组中的含量
为了准确测定在cmd1催化下5mc产生的两种衍生物的分子量,本发明人将这两种衍生物通过hplc分离,紧接着通过uplc-ms/ms进行分析测定。本发明人所使用的仪器和分析柱分别是qexactive质谱仪(thermoscientific)和acquityuplchsst3分析柱(1.8μm),使用缓冲液a(0.05%ch3cooh)和缓冲液b(乙腈,acn)作为流动相,以0.3ml/min的流速进行分析。分析时,缓冲液a和b按下列梯度进行混合,起始条件为100%a,在0-2min以线性梯度调整到95%a,2-4min以线性梯度降低到50%a,并以此混合比例运行1min,之后在0.1min内将a下降到0%,在第8min时调整回起始条件,并平衡1min。
本发明人使用基于mrm(multiplereactionmonitoring)的lc-ms/ms来检测p修饰在衣藻基因组dna中的含量。该质谱用偶联三重四级杆质谱仪(agilent6495qqq,agilenttechnologies)的uplc系统(1290series,agilenttechnologies)完成,所用分析柱为acquityuplcbehamidecolumn(1.7μm,2.1mm×100mm,waters),所用缓冲液为缓冲液a(25mm醋酸铵,25mm氨水)和缓冲液b(乙腈),以0.6ml/min的流速进行分析。分析时,缓冲液a和b按照下列梯度进行混合,起始条件为85%b,在0–2min以线性梯度降低到40%b,并以此混合比例运行2min,之后在0.1min内将b恢复到起始条件,并以此混合比例平衡3min。根据p、5mc和c的标准品优化这三种核苷在基因组dna样品中检测的测定参数,p:332.1/216.1(quantifiertransition,ce24)和332.1/150.0(qualifiertransition,ce44);5mc:242.1/126.1(quantifiertransition,ce8)和242.1/54.3(qualifiertransition,ce60)以及c:228.1/112.1(quantifiertransition,ce8)和228.1/41.3(qualifiertransition,ce64);g:268.1/152.1(quantifiertransition,ce21);268.1/135(quantifiertransition,ce45)。所有化合物均在阳离子esi模式下检测。在上述检测条件下,p、5mc和c的保留时间分别是1.50min、1.06min和1.12min。每种核苷的含量根据其在特定离子对转变(p:332.1/216.1、5mc:242.1/126.1和c:228.1/112.1、g:268.1/152.1)下测得峰面积并代入到标准曲线中计算得到。
8、核磁共振法(nmr)解析p修饰核苷结构
纯化后的p1和p2分别溶解在磷酸盐缓冲液中(0.1m,溶剂为d2o,pd7.4),并通过配备5-mmcryogenictciprobe的bruker600mhz和850mhz光谱仪采集相应的nmr数据。根据之前的文献报道(h.liuetal.,identificationofthreenovelpolyphenoliccompounds,origaninea-c,withuniqueskeletonfromoriganumvulgarel.usingthehyphenatedlc-dad-spe-nmr/msmethods.jagricfoodchem60,129-135(2012)),本发明人收集了一维1hnmr光谱和一系列二维(2d)nmr光谱数据,包括1h-1hcosy、1h-1htocsy、1hjres、1h-13chsqc和1h-13chmbc2d光谱。1h和13c的化学位移信号以tsp(δh0.000,δc0.00)的甲基信号作为参比。
根据侧链上的手性碳原子(c8和c9)及其相邻碳原子(c7和c10)上的质子计算得到三键j耦合常数(3j)。就p1和p2而言,根据在wb97xd/6-311g(d,p)水平优化后的分子几何构型密度泛函理论(dft)可以计算得到四种可能构型(8r,9r、8s,9s、8r,9s、8s,9r)的三键j耦合常数。所有结果都是根据拉姆齐理论(m.j.frischetal.(gaussian,inc.,wallingford,ct,usa,2009))在考虑了费米接触、反磁自旋轨道、顺磁自旋轨道和旋转-偶极因素下利用gaussian09软件包计算得到的。
9、莱茵衣藻(chlamydomonasreinhardtii)品系及培养方法
野生型衣藻品系(cc124/125、cc1009/cc1010、cc620/cc621)获自衣藻遗传中心(chlamydomonasgeneticcenter)。所有衣藻品系都在25℃光强为50μmol·m-2·s-1条件下在tris/acetate/phosphate(tap)培养基中振荡培养。
在5-aza-2’-deoxycytidine(5-aza,sigma)处理实验中,本发明人使用cc125衣藻品系,起始细胞浓度为1.2×104ml-1培养在含400μm5-aza的tap培养基中。在第二天更换含有新配制5-aza的tap培养基,于第四天收细胞并用于后续实验。
10、利用artificialmicrorna(amirna)在莱茵衣藻中降低cmd1表达水平
cmd1的amirna设计和构建策略参考于2014年hu等人的报道(j.hu,x.deng,n.shao,g.wang,k.huang,rapidconstructionandscreeningofartificialmicrornasystemsinchlamydomonasreinhardtii.plantj79,1052-1064(2014))。本发明人设计了八条短的寡核苷酸链(表1)用于组装产生cmd1amirna前体(该前体序列为:atcaggaaaccaaggcgcgctagcttcctgggcgcagtgttccagctgcagtacggggtccttattcaaggcatatctcgctgatcggcaccatgggggtggtggtgatcagcgctatatggcttgaataaggacccctactgcagccggaacactgccaggagaatt(seqidno:10))。之后用此前体取代phk226载体(j.hu等)上的vipp1amirna前体,得到的新质粒命名为phk227。从衣藻基因组中扩增得到rbcs2基因的第三个内含子(genbank登录号nw_001843932.1中第37072-37301位)并插入到用ecorv酶切后的phk227中,构建的新质粒命名为phk228。质粒phk228用saci线性化并用bio-radgenepulserii电转到cc125衣藻品系细胞中(k.shimogawara,s.fujiwara,a.grossman,h.usuda,high-efficiencytransformationofchlamydomonasreinhardtiibyelectroporation.genetics148,1821-1828(1998))。挑取巴龙霉素抗性克隆,并将其培养在透明的96孔板上,测定每个克隆中的荧光素酶表达水平。
表1、合成靶向cmd1的amirna前体序列
为了进一步测定cmd1在细胞内的表达水平,本发明人用qpcr(quantitativepcr)测定上述筛选到的高荧光素酶活性的细胞克隆中的cmd1mrna的转录水平,所用仪器为cfxp6tmreal-timepcr仪,所用试剂为sybrpremixtmextaq(tlirnasehplus,takara)。使用引物cmd1-qf(5′-ccacaaacatcatctctctcacc-3′(seqidno:13))和cmd1-qr(5′-atggtgaagtccagtggttga-3′(seqidno:14))来扩增cmd1cdna片段,使用引物cblp-qf(5′-gtcatccactgcctgtgctt-3′(seqidno:15))和cblp-qr(5′-ccttcttgctggtgatgttg-3′(seqidno:16))来扩增cblp(g-proteinbetasubunit-likepolypeptide)cdna片段。cblp在本实验中被用作内参。
11、莱茵衣藻基因组dna的大量制备
ctab被用来抽提莱茵衣藻的基因组dna,并溶于无核酸酶的水中用于后续实验。
12、统计学分析
所有实验数据都是通过三次独立重复实验计算得到的平均标准误,每组数据的统计学差异都是利用graphpad软件中的双尾检验进行评估。当p<0.05时,就认为存在显著性差异。
实施例1、cmd1催化5mc生成2种新修饰
为了研究衣藻tet同源蛋白crtet的酶活,本发明人从大肠杆菌中表达、纯化出了这些蛋白并进行了体外酶活分析。从大肠杆菌中纯化到的不含标签的cmd1的考马斯亮蓝染色图如图2a;cmd1突变体的考马斯亮蓝染色图如图3a。
出乎意料的是,以5mc-dna为底物进行crtet1酶活实验测试之后,hplc结果显示,有2个与核苷标准品具有完全不同的保留时间的峰(p1/p2)产生,而利用crtet1的保守位点的突变蛋白(mutcmd1,h345/d347)进行测试时则无法检测到相应的峰;同时,5mc-dna与cmd1反应,随着时间增加,p1/p2含量的积累与5mc的减少是一致的,如图2b-d、图3a-b。
然而,本发明人在利用5hmc-dna以及c-dna(指常规的未修饰dna)作为底物进行反应的时候,却无法检测到p1/p2的积累,说明这个反应很有可能以5mc作为直接底物,如图3c。
为进一步确定hplc的结果,本发明人用14c放射性同位素标记5mc-dna的甲基碳原子,并用于薄层层析(tlc)实验。以该同位素标记5mc-dna为底物,经过与野生型cmd1反应之后,在层析板上本发明人同样观测到了2个未知产物形成的点,而某些保守氨基酸位点的突变型cmd1没有该功能,如图2e。
以上实验均表明,本发明人发现了一种新的5mc修饰酶(cmd1),可以催化5mc生成2种新修饰。并且,根据实验结果以及序列比对结果分析,提示h345和d347是与亚铁离子结合的位点,a330是cmd1的酶活中心位点,d350与5mc相互作用。也即a330、h345、d347和d350是cmd1发挥酶活的关键性位点。cmd1有500多个氨基酸,a330、h345、d347和d350这些保守位点以外的其它一些非保守位置的氨基酸有可能可以被相近的氨基酸替换。
实施例2、解析p1、p2两种新产物的结构
为了解析实施例1发现的两种新产物的结构,本发明人将分离出的反应产物进行高分辨质谱分析。
质谱结果显示,在正离子模式下,p1、p2的质荷比分别是332.1448、332.1449,以此推测出p1/p2的分子式同为c13h21n3o7,如图4a。
由于p1、p2在二级质谱中的碎片离子谱式完全一致,可以推测p1、p2属于立体异构体,如图5c。
为了确定p1、p2相比于5mc所增加的90道尔顿的分子量是否直接添加在甲基上,氘代5mc-dna被用于cmd1酶活实验。相比未标记的5mc来说,氘代5mc的分子量增加3,如图5a。而利用氘代5mc经过cmd1反应产生的p1/p2,分子量与未标记的相比仅增加2,如图5b。这说明5mc的甲基基团上的一个氢原子在cmd1的酶活反应中被取代,且剩余的亚甲基在二级质谱的碎片中都得到保留。仔细分析二级质谱的分子碎片本发明人可以发现,p1/p2离子在中性丢失一个脱氧核糖之后,又连续失去了3个水分子,暗示在p1/p2的分子结构上可能存在3个羟基,如图5c。
为了确定p1/p2的分子结构,本发明人进行了核磁共振实验以及密度泛函理论计算。二维核磁谱图较为清晰地给出了p1/p2的平面分子结构。以p1为例,cosy与tocsy的峰证实了三羟基丁基,-ch2ch(oh)ch(oh)ch2oh基团的存在。而hsqc与hmbc谱图进一步确认了不同原子之间的连接关系,并证实p1的结构式为:5-(1-[2,3,4-三羟基丁基])-2′-脱氧胞嘧啶。为了证明其中2个手性碳原子(c8,c9)的绝对构型,根据质子的平面位置以及键角所计算出来的理论j耦合常数被用来与实验测定值相比较(m.karplus,vicinalprotoncouplinginnuclearmagneticresonance.journaloftheamericanchemicalsociety85,2870-2871(1963)),p1被推断出具有(8s,9s)构型,而p2具有(8r,9s)构型。
因此,根据二维核磁共振以及密度泛函理论所确定的p1/p2的结构式如图4b。p1与p2是立体异构体,仅在c8位置存在手性差异。
实施例3、5mc上的甘油基的来源分析
为了探究添加到5mc上的甘油基的来源,本发明人首先想确认其是否来自于cmd1共纯化的蛋白或小分子化合物。利用13c-标记的葡萄糖作为唯一碳源来培养大肠杆菌之后,cmd1被纯化出来进行酶活反应。将获得的反应产物p1/p2进行质谱分析后,本发明人发现其分子量并没有增加,说明甘油基来自于体外酶活反应体系(图6a)。在针对反应体系中的成分进行筛选之后,本发明人发现维生素c(vc)在cmd1酶活反应中是十分必需的,而不仅是在传统的tet等双加氧酶实验中起到一个还原剂并促进酶活的作用(图6b、6d)。另一方面,2-og作为许多双加氧酶所必需的辅因子,在cmd1的酶活反应中却可有可无(图6b、6c)。因此本发明人检测了利用全部六个碳都用13c标记的vc反应产生的p1/p2的分子量,发现其与原来相比增加了3。进一步用单个碳原子标记13c的vc进行实验后发现,vc上c4-c6直接转移到5mc上形成了新的修饰(图7)。且vc上的c-6位碳原子对应的是p1上的c-10,vc上c-5对应的是p1的c-9。而利用vc的类似物,包括d-异抗坏血酸等,都无法直接替代vc进行酶活实验。这些实验结果都表明vc作为一个直接的反应底物参与cmd1的酶活反应(6e-g)。值得一提的是,亚铁离子fe2+在cmd1的反应中也是必需的(图6h)。但是,vc的某些类似物也能作为直接或间接的反应底物参与cmd1的酶活反应,发挥与vc相同的效果,这些类似物包括:脱氢抗坏血酸(其经dtt还原来间接参与反应)等(图6e)。
接下来本发明人检测了氧气在cmd1酶活反应中的功能,在充满氮气的无氧手套箱中进行实验,发现在无氧条件下,cmd1的酶活被抑制。而如果往反应瓶中鼓入氧气,则cmd1的酶活可以正常发挥(图8a)。但氧气与水中的氧原子并没有被转移到反应产物p1中,进一步说明p1中增加的原子组成都来自vc(图8b)。
参考传统的双加氧酶的反应原理,本发明人也提出了预期的cmd1的反应机理(图8c)。在这个模式图中,vc替代了2-og与fe(ii)进行配位,其经过氧化脱羧形成的活性基团攫取了5mc的甲基氢原子,形成的5mc自由基又造成了fe(ii)与羧酸的配位中间体的均裂,最终形成了p修饰核苷以及副产物乙醛酸。为了验证这个反应机理,本发明人首先需要检测vc的反应副产物的产生。利用气相色谱(gc-ms)实验,本发明人证实了在利用13c-vc进行反应后,co2由vc的c-1位产生(图8d)。为了检测乙醛酸的生成,本发明人在cmd1的反应产物中加入2,4-二硝基苯肼(2,4-dinitrophenylhydrazine,dnp)进行衍生,并利用lc-ms检测其衍生产物。在实验过程中,本发明人利用13c-vc,发现vc中的c-2,c-3转变成了乙醛酸(图8e)。
实施例4、体内酶活反应
本发明人已经证明在体外酶活反应中,cmd1催化5mc生成p1/p2两种新修饰,本实施例验证这个反应过程在衣藻细胞基因组中同样发生。
利用三重四级杆质谱仪,本发明人测试了六株不同品系的衣藻藻株基因组dna碱基组成。在这些藻株中都可以检测到p修饰碱基的存在,大约占c的比例的百万分之10-35,占5mc的0.2%左右(图9a,9b)。
为了证实p修饰碱基在衣藻体内同样也是由cmd1催化完成,本发明人制备了cmd1knockdown的藻株。利用人工制备的microrna(amirna),得到了几株knockdown效率在70%-80%的藻株(图10)。
质谱结果显示,在这些藻株的基因组中,p修饰碱基的含量大约降低了35%,而相对应的5mc的含量增加了17%左右(图9c,9d)。当用5-azacytidine这个甲基转移酶的抑制剂来培养衣藻之后,本发明人发现5mc在基因组中的含量降低了50%以上,而相一致的是,p修饰碱基的含量同样降低了13%左右,表明在基因组中,p修饰碱基同样来源于5mc(图9e,9f)。
实施例5、vc衍生的5mc新修饰(p修饰)的功能与单碱基水平检测
1、报告基因表达检测显示p修饰抑制基因表达
利用荧光素酶报告基因的方法,本发明人发现p修饰能强烈抑制报告基因的表达。采用pcpgl-cmv-firefly报告质粒,该质粒仅在cmv启动子区域含有cpg位点,本发明人利用m.sssi甲基转移酶对报告质粒进行体外甲基化后,就可以获得在cmv启动子区域高度甲基化的报告质粒。更进一步用cmd1酶处理后,获得在cmv启动子区域含p修饰的报告质粒。通过在hek293t细胞中转染这个报告质粒,并与未修饰的普通质粒,以及5mc报告质粒进行对比。本发明人发现,含p修饰的报告质粒的基因表达受到明显抑制,且无法被tet与tdg介导的dna主动去甲基化途径激活,如图12a。
2、p修饰的单碱基检测
为了研究vc衍生的新修饰在基因组中的位点特异性分布,以及其潜在的功能,并同时考虑了对于分析核酸分子中各种胞嘧啶修饰的需求,本发明人设计了围绕p修饰的单碱基检测手段,如图12b、c、d。
如图12b所示,这是一种tet双加氧酶氧化耦联的bisulfite-seq方法,即首先用tet双加氧酶将dna中的5mc,5hmc以及5fc全部氧化成5cac,而p修饰因无法被tet氧化而不能在bisulfite处理时发生脱氨,因此经bisulfite-seq就可以明确地区分出5mc(以t的形式出现)与p修饰(以c的形式出现)。
在dna中不存在p修饰的情况下,利用以下方法能区分5mc与5hmc:首先,用cmd1对dna进行处理,将5mc转变为p修饰,而5hmc仍保持原样。再进一步利用tet双加氧酶将5hmc氧化成5cac(5cac可被bisulfite脱氨转变),从而将5mc与5hmc区分开来。
p修饰碱基在bisulfite-seq中的模式与5mc一样,而由于5mc上连接了甘油基,因此无法像5mc那样被tet双加氧酶进一步氧化成可以发生脱氨反应的碱基,因此可以用这个tet-oxidizedbisulfite-seq(tet-oxi-bs-seq)的方式来区分p修饰与5mc。
cmd1催化5mc与vc反应生成新的修饰产物的特性,可被用在最新的dna测序技术中检测5mc的含量与分布。
3、p修饰对于酶切割的影响
酶切实验还发现,cmd1介导的p修饰影响了限制性内切酶mspi(识别ccgg序列)对5mc-dna的切割活性。mspi可切割含5mc修饰的dna,但经过cmd1反应转变成p修饰之后,mspi的功能被阻遏,如图13。
讨论
如前所述,本发明人发现衣藻的一个tet同源蛋白(cmd1)可以催化基因组中的5mc与vc上的c4-c6组成的甘油基发生反应生成新的dna修饰。作为多种生命体中十分重要的营养物质,vc被认为在多种生理过程中承担着还原剂的作用,例如在表观遗传修饰重编程过程中通过维持fe2+的稳定性等方式促进tet与组蛋白去甲基化酶的酶活(m.a.esteban,d.pei,vitamincimprovesthequalityofsomaticcellreprogramming.naturegenetics44,366-367(2012);j.du,j.j.cullen,g.r.buettner,ascorbicacid:chemistry,biologyandthetreatmentofcancer.bba-revcancer1826,443-457(2012))。vc的c4-c6及其链上的羟基的转移涉及到c-c单键的断裂和重构,这类反应在有机化学上也从未被报道过(图11)。虽然所涉及的化学机制还不完全清楚,但至少可以确定这和传统的双加氧酶介导的氧化反应机理存在较大差异,尤其是2-og在cmd1反应中并不起作用,而且cmd1蛋白序列上也不存在2-og的结合位点。在更广义的依赖于fe2+/2-og的双加氧酶家族中,合成乙烯的酶,acc氧化酶已经被证实需要vc,而不是2-og作为还原剂。未来对vc的反应副产物的检测以及fe2+所起到的作用的了解可以帮助本发明人更好地理解反应机制。本发明人的实验也预示着vc存在不为人知的除了氧化还原调控之外的新功能,并暗示存在dna修饰与代谢产物相互影响的可能性。
vc反应产生的dna新修饰在衣藻中的功能也有待研究。但至少本发明人从cmd1knockdown的藻株中观测到的5mc水平升高,暗示cmd1可能起到了调控5mc水平的作用,恰如tet双加氧酶在哺乳动物中的角色。更进一步的质谱分析结果显示,包括哺乳动物在内的其它几种与莱茵衣藻在进化上相近的生物体可能并不存在这种p修饰碱基或修饰水平很低。利用荧光素酶报告基因的方法,本发明人发现p修饰至少在哺乳动物细胞中能强烈抑制报告基因的表达(图12a)。
为了研究vc衍生的新修饰在基因组中的位点特异性分布,以及其潜在的功能,本发明人设计了围绕p修饰的单碱基检测手段(图12b、c、d)。p修饰碱基在bisulfite-seq中的模式与5mc一样。而当5mc上连接了甘油基,就无法被tet双加氧酶进一步氧化,因此可以用tet-oxidizedbisulfite-seq这个的办法来区分p与5mc修饰。当然,cmd1催化5mc与vc反应生成的新产物也可能被用在最新的dna测序中检测5mc的含量。
另外,cmd1酶活所涉及的反应机理也可促进对现有的有机化学理论进一步的理解和探讨,有助于新的有机化学实验,包括碳链的延伸,多羟基的添加等方案的设计。进一步的研究也可能促进本发明人对cmd1蛋白的改造,进而达到在细胞内调控基因表达,设计特异性表观遗传药物等目标。
在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
序列表
<110>中国科学院上海生命科学研究院
<120>cmd1酶催化的5-甲基胞嘧啶dna的新修饰及其应用
<130>165231z1
<150>201610719692.4
<151>2016-08-24
<160>16
<170>siposequencelisting1.0
<210>1
<211>532
<212>prt
<213>莱茵衣藻(chlamydomonasreinhardtii)
<400>1
metservalalaleualaserglutyrglnleuvalglnasnalagln
151015
leuproglnargtrpserglnseralaarglysserleualaileleu
202530
glualathralaarglysglualathralaglnmetglualaalagly
354045
glyserphecysglyglnpheprovalaspproalaphelysvalleu
505560
serleuglutyrseralaproasnproaspilealaargalailearg
65707580
argvalaspservalproasnproproleuproserhisvalvalala
859095
ileglnserthralavalaspalaaspleuserleualametglyval
100105110
serleuthrproglyarghisthrsertyrleuvalaspalaargala
115120125
leuglnglnserasnseralaalavalalaalaarglysalaaspgly
130135140
asplystrpglyproalacysaspglumetpheargglycysargcys
145150155160
valthrglyglngluvalvalphetyrthralavallysgluproala
165170175
glygluvalgluglyglygluglyserleuphelysproserpheasp
180185190
glyproalapheargprosertrpglygluleuserglylysalathr
195200205
glyvalvalalacysvalleuglnvalproileglylysgluthrasp
210215220
ileilecysalaglutyraspasnleuvalserlysglyglnpheala
225230235240
thrvalaspargpheglyglyasphisthrvalasnmetthrglyasn
245250255
alaleuileglnasnaspglylysalaileserlysglytyralaval
260265270
alahisargalaargvalthrserasnvaltyrglylysalaasnasp
275280285
valserleuglnargleualagluthrvaltrpservalvalglulys
290295300
argleuserphemetproalatyrargaspleuvalilethrglugln
305310315320
glylysprophemetleuglyalathralathrasnileileserleu
325330335
thrgluasnglnglyvalmetleuhisleuaspthraspaspglyval
340345350
trpthrileileleutrpphehisarghisserglyileilealagly
355360365
glygluphevalleuproserleuglyileserpheglnproleuasp
370375380
phethrilevalvalphealaalaasnthrilevalhisglythrarg
385390395400
proleuglnthrthrglylysileileargtrpglyserserhisphe
405410415
leuargphelysaspvalasnalaleualaglnleuglyalaalatyr
420425430
glyvalaspgluleuaspalalysglnargaspglnleuglugluval
435440445
aspalaalaasnserlysaspglyvalglyalaalaargargvalala
450455460
sercysmetalaalagluarglysalaalaileglualaglnlysala
465470475480
alacysvalargglyvalvalmetasnprocysthrglyargmetpro
485490495
serleuleuphetrpglnvaltrparglysproproalaleualaval
500505510
argalaasnalavalalaglylyslysargalaalaalaaspvalasp
515520525
phecysglyala
530
<210>2
<211>25
<212>dna
<213>人工序列(artificalsequence)
<220>
<221>misc_feature
<222>(1)..(25)
<223>多核苷酸
<400>2
atcaggaaaccaaggcgcgctagct25
<210>3
<211>54
<212>dna
<213>人工序列(artificalsequence)
<220>
<221>misc_feature
<222>(1)..(54)
<223>多核苷酸
<400>3
gtactgcagctggaacactgcgcccaggaagctagcgcgccttggtttcctgat54
<210>4
<211>50
<212>dna
<213>人工序列(artificalsequence)
<220>
<221>misc_feature
<222>(1)..(50)
<223>多核苷酸
<400>4
tcctgggcgcagtgttccagctgcagtacggggtccttattcaaggcata50
<210>5
<211>42
<212>dna
<213>人工序列(artificalsequence)
<220>
<221>misc_feature
<222>(1)..(42)
<223>多核苷酸
<400>5
ccatggtgccgatcagcgagatatgccttgaataaggacccc42
<210>6
<211>42
<212>dna
<213>人工序列(artificalsequence)
<220>
<221>misc_feature
<222>(1)..(42)
<223>多核苷酸
<400>6
tctcgctgatcggcaccatgggggtggtggtgatcagcgcta42
<210>7
<211>42
<212>dna
<213>人工序列(artificalsequence)
<220>
<221>misc_feature
<222>(1)..(42)
<223>多核苷酸
<400>7
ggggtccttattcaagccatatagcgctgatcaccaccaccc42
<210>8
<211>47
<212>dna
<213>人工序列(artificalsequence)
<220>
<221>misc_feature
<222>(1)..(47)
<223>多核苷酸
<400>8
tatggcttgaataaggacccctactgcagccggaacactgccaggag47
<210>9
<211>30
<212>dna
<213>人工序列(artificalsequence)
<220>
<221>misc_feature
<222>(1)..(30)
<223>多核苷酸
<400>9
aattctcctggcagtgttccggctgcagta30
<210>10
<211>168
<212>dna
<213>人工序列(artificalsequence)
<220>
<221>misc_feature
<222>(1)..(168)
<223>多核苷酸
<400>10
atcaggaaaccaaggcgcgctagcttcctgggcgcagtgttccagctgcagtacggggtc60
cttattcaaggcatatctcgctgatcggcaccatgggggtggtggtgatcagcgctatat120
ggcttgaataaggacccctactgcagccggaacactgccaggagaatt168
<210>11
<211>29
<212>dna
<213>人工序列(artificalsequence)
<220>
<221>misc_feature
<222>(1)..(29)
<223>引物
<400>11
aagggttggattgtaggtagtttagaaat29
<210>12
<211>17
<212>dna
<213>人工序列(artificalsequence)
<220>
<221>misc_feature
<222>(1)..(17)
<223>引物
<400>12
tgagggtggtaaattag17
<210>13
<211>23
<212>dna
<213>人工序列(artificalsequence)
<220>
<221>misc_feature
<222>(1)..(23)
<223>引物
<400>13
ccacaaacatcatctctctcacc23
<210>14
<211>21
<212>dna
<213>人工序列(artificalsequence)
<220>
<221>misc_feature
<222>(1)..(21)
<223>引物
<400>14
atggtgaagtccagtggttga21
<210>15
<211>20
<212>dna
<213>人工序列(artificalsequence)
<220>
<221>misc_feature
<222>(1)..(20)
<223>引物
<400>15
gtcatccactgcctgtgctt20
<210>16
<211>20
<212>dna
<213>人工序列(artificalsequence)
<220>
<221>misc_feature
<222>(1)..(20)
<223>引物
<400>16
ccttcttgctggtgatgttg20
- 还没有人留言评论。精彩留言会获得点赞!