一种盐酸维拉佐酮IV晶的制备方法与流程
本发明涉及一种盐酸维拉佐酮晶型的制备方法,具体地说,涉及盐酸维拉佐酮iv晶的制备方法。
背景技术:
维拉佐酮片在2011年被美国fda批准用于治疗成人重度抑郁症,是首个吲哚烷基胺类新型抗抑郁药,双重作用机制:选择性5羟色胺再摄取抑制剂和5-ht1a受体部分激动剂,起效时间明显优于现有的ssris类抗抑郁药物,从而大大增加了患者的依从性。
同时维拉佐酮的临床数据试验表明,其疗效明显优于安慰剂,具有起效快、响应率高、顺应性好、不增加体重,也不像许多抗抑郁药那样影响性功能的优点,而且不良反应小,其常见(发生率≥5%和至少是安慰剂发生率的2倍)的不良反应为腹泻、恶心、呕吐和失眠。
经研究,盐酸维拉佐酮为多晶药物,而不同晶型在外观、溶解度、熔点、溶出度、生物有效性等方面会有显著不同,从而影响了药物的稳定性、生物利用度及疗效,这种现象在口服固体制剂方面表现得尤为明显。
专利zl02812226.7(cn100384841c)公开了维拉佐酮盐酸盐的各种晶型:1)1-[4-(5-氰基吲哚-3-基)丁基]-4-(2-氨甲酰-苯并呋喃-5-基)-哌嗪盐酸盐的溶剂化物,晶型i:丙酮化物;晶型ii和xv:与四氢呋喃的一溶剂化物;晶型x:与四氢呋喃的半溶剂化物;晶型xi:甲醇化物;晶型xiv:与正庚烷的一溶剂化物;2)1-[4-(5-氰基吲哚-3-基)丁基]-4-(2-氨甲酰-苯并呋喃-5-基)-哌嗪盐酸盐的水合物,晶型v:一水合物;晶型vi:倍半水合物;晶型viii:半水合物;3)1-[4-(5-氰基吲哚-3-基)丁基]-4-(2-氨甲酰-苯并呋喃-5-基)-哌嗪盐酸盐的脱水物,晶型iv、iii、vii、ix;4)1-[4-(5-氰基吲哚-3-基)丁基]-4-(2-氨甲酰-苯并呋喃-5-基)-哌嗪二盐酸盐,晶型xiii。
其中,在较高的温度下,例如>100℃,iv型是最稳定的形态;据描述,iv晶在溶解度和在药学加工成固体制剂方面均有一定优势,同时iv型在水中的溶解度为0.328μg/ml,是无色固体物质,为充分确定的晶型形式,是优势药物晶型。
在维拉佐酮片原研默克公司申报的此专利中,发明人经过探索指出,“这种特定的多晶型物形态(本文成为“iv晶”)具有优于其他结晶形态的性质,更适合于包括在药物制剂中”。
结合zl200710180229.8(cn101139345b),可知iv晶的xrd数据为:
专利zl02812226.7(cn100384841c)中公开的iv晶的合成方法,包含:1)将1-[4-(5-氰基吲哚-3-基)丁基]-4-(2-氨甲酰-苯并呋喃-5-基)-哌嗪分散在四氢呋喃中;2)在20℃与30℃之间的温度下加入含水盐酸,转化1-[4-(5-氰基吲哚-3-基)丁基]-4-(2-氨甲酰-苯并呋喃-5-基)-哌嗪为盐酸盐;3)在室温下沉淀出v晶;4)借助过滤回收所沉淀的1-[4-(5-氰基吲哚-3-基)丁基]-4-(2-氨甲酰-苯并呋喃-5-基)-哌嗪盐酸盐一水合物v晶;5)在85至90℃真空中干燥v型,得到iv晶。
或者在55与65℃之间的温度下干燥xi型1-[4-(5-氰基吲哚-3-基)丁基]-4-(2-氨甲酰-苯并呋喃-5-基)-哌嗪盐酸盐一甲醇化物,得到iv晶。
上述两种iv晶型制备方法的共同点是通过盐酸维拉佐酮的其他晶型,经不同温度条件下真空干燥转晶而得,这非常容易导致混晶现象的发生,且相关iv晶型制备工艺比较复杂。比如:原研v晶在85-90℃真空干燥得到iv晶,而众所周知,固体状态下的转晶会有包裹在颗粒中不完全转晶的情况发生。同时,在上述专利申请文件中要求保护含iv晶和v晶的组合物或者混合物,二者的摩尔比为约100比1至10比1。
现有技术中,默克专利有限公司申请的盐酸维拉佐酮iv晶的制备方法,在iv晶的制备过程中使用了含水盐酸,从水中析晶,将导致盐酸维拉佐酮含水量高,经研究,高含水量对于盐酸维拉佐酮片的溶出度产生了不利影响,降低了药物的稳定性、生物利用度及疗效。
因此急需一种新的盐酸维拉佐酮iv晶的制备方法,克服现有技术中盐酸维拉佐酮iv晶和v晶发生混晶,及产生的盐酸维拉佐酮含水量对制剂溶出度产生不利影响的缺陷。
技术实现要素:
为了解决现有技术的上述技术问题,本发明提供了一种新的盐酸维拉佐酮iv晶的制备方法,这种方法简单、便捷,出人意料地解决了现有技术中iv晶通过其他晶型固态下转晶所引起的混晶现象,避免了混合物的产生;生成的盐酸维拉佐酮iv晶粒度适宜,残留溶剂合格,并且含水量低,有利于得到稳定的晶型并改善制剂产品的溶出情况,绘制令人满意的体外溶出曲线,可提升药物稳定性、生物利用度及疗效。
本发明的目的是提供一种盐酸维拉佐酮iv晶的制备方法,该制备方法包括以下步骤:
(1)将1-[4-(5-氰基吲哚-3-基)丁基]-4-(2-氨甲酰-苯并呋喃-5-基)-哌嗪加入异丙醇中,加热回流后过滤;
(2)向滤液中滴加含5%~35%氯化氢的异丙醇溶液;
(3)滴加完毕后回流,过滤,烘干得到iv晶。
在上述制备方法中,通过将1-[4-(5-氰基吲哚-3-基)丁基]-4-(2-氨甲酰-苯并呋喃-5-基)-哌嗪溶解于异丙醇溶剂中,使用含5%~35%氯化氢的异丙醇溶液,在不含水的条件下生成维拉佐酮盐酸盐,直接可以得到iv晶。该方法简单、便捷,不通过v晶或xi晶转晶,尤其避免了固态条件下转晶容易混晶的缺陷,同时使用了不含水的环境,生成的维拉佐酮盐酸盐含水量低,有利于得到稳定的晶型,并改善制剂产品的溶出曲线,对于药物稳定性、生物利用度、疗效均所提升。
同时发明人发现,反应中滴加的含氯化氢的异丙醇溶液中,所含氯化氢的浓度对于反应体系有较大影响。如果氯化氢异丙醇溶液浓度过低,比如低于5%,会造成析出晶体过大,并包裹了部分异丙醇溶剂,不但造成残留溶剂不合格,较难通过后续干燥除去,而且不能达到后续制剂所需的原料粒度,造成溶出度不合格,此时需要对原料进行后续粉碎,增加操作成本,并容易造成产品污染。而采用5%~35%氯化氢的异丙醇溶液,可以基本解决上述问题。
优选地,在步骤1)中,异丙醇与1-[4-(5-氰基吲哚-3-基)丁基]-4-(2-氨甲酰-苯并呋喃-5-基)-哌嗪的体积/质量比(ml/g)为50~150;其中更优选为70~100。
经过发明人的探索,发现异丙醇与1-[4-(5-氰基吲哚-3-基)丁基]-4-(2-氨甲酰-苯并呋喃-5-基)-哌嗪的体积/质量比(ml/g)对于反应收率和维拉佐酮盐酸盐的纯度及有关物质情况均有较大影响。经过不断探索发现,该体积/质量比(ml/g)优选为在50~150内,最佳在70~100,避免维拉佐酮游离碱在反应过程中,从反应体系中析出,将提高反应收率和最终产品的纯度,制备合格的最终产品。
优选地,在步骤1)中,加热回流时间为0.5~2小时,更优选加热回流时间为0.5~1小时,过滤之后,将滤液加热至50~75℃,更优选将所述滤液加热至65~75℃。
将1-[4-(5-氰基吲哚-3-基)丁基]-4-(2-氨甲酰-苯并呋喃-5-基)-哌嗪加入异丙醇中,控制加热回流的时间,可以保证其充分溶解于异丙醇中,小于0.5小时,容易溶解不完全,超过2小时也会造成成本的浪费;过滤掉不溶物后,将滤液加热至特定的温度,可以保持维拉佐酮游离碱继续溶解于滤液中。
优选地,在步骤2)中,含氯化氢的异丙醇溶液,浓度为10%~15%。
市场上常见的氯化氢的异丙醇溶液,浓度大约是25~35%,大约为6.85~9.60n。经过发明人深入研究,发现浓度大于15%,滴定液使用量将大幅减少,产品粒度比较小,但是会造成析晶过快,也容易产生异丙醇包裹,一方面较难控制滴定速度,另一方面造成产品粒度不均一,因此发明人将市售的氯化氢异丙醇溶液稀释后使用,使用时的最佳浓度在10%~15%。
优选地,在步骤2)中,滴加过程保持温度在50~75℃,优选为65~75℃。
滴加含氯化氢的异丙醇溶液的过程中,需要控制滴加温度,适宜的滴加温度,一方面保证了反应速度;另一方面保证了未反应的1-[4-(5-氰基吲哚-3-基)丁基]-4-(2-氨甲酰-苯并呋喃-5-基)-哌嗪溶解于异丙醇中,有利于反应的进行,不会与维拉佐酮盐酸盐同时析出,影响产品纯度;第三,经研究,在此温度下维拉佐酮盐酸盐的析出速度适当,析晶颗粒大小适宜,包裹较少的异丙醇溶剂,使得最终产品的纯度、粒度和残留溶剂量满足制剂的制备需要。
优选地,在步骤2)中,滴加过程持续0.2~2小时,更优选为0.5~1小时。
同样的,滴加过程的持续时间也会影响到析晶速度、产品粒度和残留溶剂含量等。滴加速度过快,容易产生异丙醇包裹,造成产品粒度不均一;滴加速度超过1小时,容易造成产品粒度过大,残留溶剂不合格,并影响后续制剂的体外溶出,需要对制备得到的iv晶产品进行粉碎操作,适宜的滴加时间可以进一步保证盐酸维拉佐酮iv晶的最终质量。
优选地,在步骤3)中,滴加完毕后回流1.5~6小时,更优选2~4小时,过滤后在80-100℃,真空度-0.09~-0.1mpa下,干燥10~25小时,更优选12~18小时。
滴加完毕后回流1.5~6小时,更优选2~4小时,可以使得反应完全,保证最终产品的纯度;一定的干燥温度和干燥时间,可以保证维拉佐酮盐酸盐iv晶具有适宜的含水量和残留溶剂,从而有利于得到稳定的晶型并改善后续制剂的溶出情况,得到令人满意的溶出曲线。
优选地,使用本发明的制备方法得到的iv晶,粒度在10~200μm,优选d90<50μm,异丙醇溶剂残留<0.5%。
在对盐酸维拉佐酮片开展体外溶出试验过程中研究发现,盐酸维拉佐酮iv晶的粒度对于口服片剂的溶出度产生较大影响,控制粒度在10~200μm,优选d90<50μm,异丙醇溶剂残留<0.5%,使用本发明的制备方法得到的iv晶,粒度适宜,溶剂残留合格,进一步制备得到的盐酸维拉佐酮片可以得到良好的溶出曲线,满足临床需要。
附图说明:
图1盐酸维拉佐酮iv晶(07)的x衍射图谱;
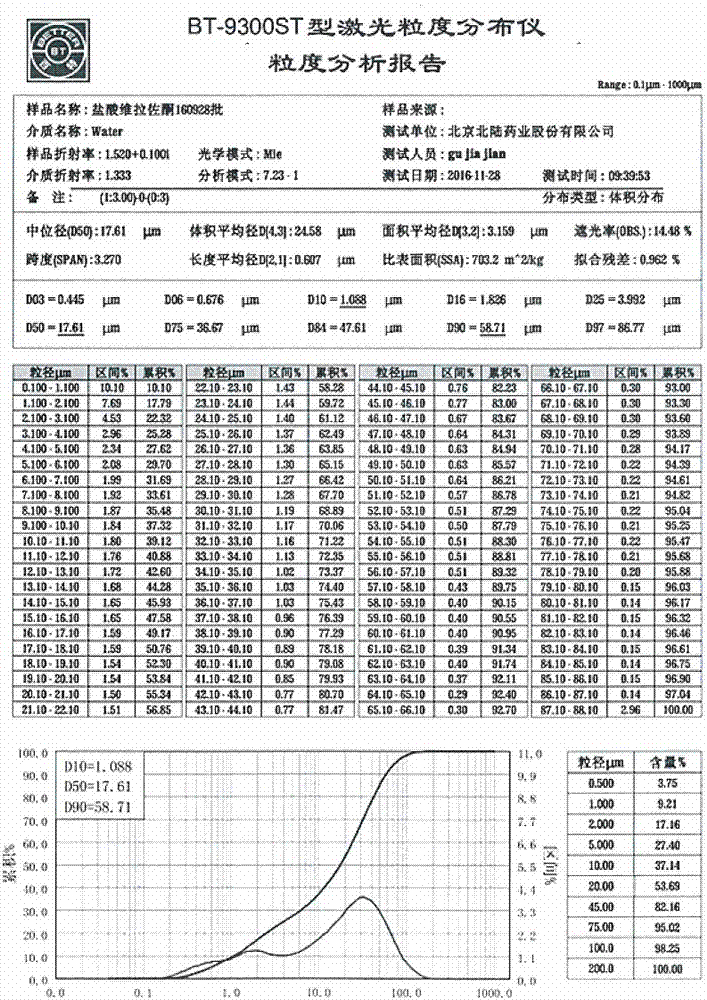
图2盐酸维拉佐酮iv晶(02)的粒度图谱;
图3盐酸维拉佐酮iv晶(07)的粒度图谱;
图4盐酸维拉佐酮iv晶(02)的残留溶剂图谱(1);
图5盐酸维拉佐酮iv晶(02)的残留溶剂图谱(2);
图6盐酸维拉佐酮iv晶(07)的残留溶剂图谱(1);
图7盐酸维拉佐酮iv晶(07)的残留溶剂图谱(2);
图8盐酸维拉佐酮iv晶(02)自制制剂的溶出曲线;
图9盐酸维拉佐酮iv晶(07)自制制剂的溶出曲线。
具体实施方式
为了进一步阐明本发明,下面给出一系列实施例。需要指出的是,这些实施例完全是例证性的。给出这些实施例的目的是为了充分明示本发明的意义和内容,但并不因此将本发明限制在所述的实施例范围之中。
对比实施例1:盐酸维拉佐酮iv晶的制备(01)
向1g1-[4-(5-氰基吲哚-3-基)丁基]-4-(2-氨甲酰-苯并呋喃-5-基)-哌嗪的32.6g四氢呋喃溶液加入2.1g盐酸(37重量%)。搅拌后,对所沉淀的晶体进行抽吸过滤。在室温真空中干燥至恒重,得到1-[4-(5-氰基吲哚-3-基)丁基]-4-(2-氨甲酰-苯并呋喃-5-基)-哌嗪盐酸盐水合物v晶。
在85至90℃真空中干燥制备的v晶至恒重,得到1-[4-(5-氰基吲哚-3-基)丁基]-4-(2-氨甲酰-苯并呋喃-5-基)-哌嗪盐酸盐iv晶0.9g,产品纯度(非晶体纯度)是95.20%,收率83.13%。
对比实施例2:盐酸维拉佐酮iv晶的制备(02)
将44g1-[4-(5-氰基吲哚-3-基)丁基]-4-(2-氨甲酰-苯并呋喃-5-基)-哌嗪加入到1500ml异丙醇中,加热至回流,回流20分钟后趁热过滤,室温下滴加含4%氯化氢的异丙醇溶液92ml,滴加过程中保持温度为45℃,持续1.5小时,滴加完毕后加热至回流,回流2小时,过滤,在80-100℃,真空度-0.09~-0.1mpa条件下烘干10小时,收料,制得盐酸维拉佐酮iv晶(01)35.7g,纯度是90.79%,收率74.95%。
实施例3:盐酸维拉佐酮iv晶的制备(03)
将44g1-[4-(5-氰基吲哚-3-基)丁基]-4-(2-氨甲酰-苯并呋喃-5-基)-哌嗪加入到2200ml异丙醇中,加热至回流,回流2小时后趁热过滤,将滤液加热到50℃,滴加含5%氯化氢的异丙醇溶液73ml,滴加过程中保持温度为50℃,持续2小时,滴加完毕后加热至回流,回流1.5小时,过滤,在80-100℃,真空度-0.09~-0.1mpa条件下烘干10小时,收料,制得盐酸维拉佐酮iv晶(01)41.0g,纯度是96.55%,收率86.07%。
实施例4:盐酸维拉佐酮iv晶的制备(04)
将44g1-[4-(5-氰基吲哚-3-基)丁基]-4-(2-氨甲酰-苯并呋喃-5-基)-哌嗪加入到3080ml异丙醇中,加热至回流,回流0.5小时后趁热过滤,将滤液加热到65℃,滴加含10%氯化氢的异丙醇溶液36.5ml,滴加过程中保持温度为65℃,持续1小时,滴加完毕后加热至回流,回流2小时,过滤,在80-100℃,真空度-0.09~-0.1mpa条件下烘干12小时,收料,制得盐酸维拉佐酮iv晶(02)43.5g,纯度是98.23%,收率91.32%。
实施例5:盐酸维拉佐酮iv晶的制备(05)
将44g1-[4-(5-氰基吲哚-3-基)丁基]-4-(2-氨甲酰-苯并呋喃-5-基)-哌嗪加入到4400ml异丙醇中,加热至回流,回流0.5小时后趁热过滤,将滤液加热到75℃,滴加含15%氯化氢的异丙醇溶液24.3ml,滴加过程中保持温度为75℃,持0.5小时,滴加完毕后加热至回流,回流4小时,过滤,在80-100℃,真空度-0.09~-0.1mpa条件下烘干18小时,收料,制得盐酸维拉佐酮iv晶(03)44.8g,纯度是99.46%,收率94.05%。
实施例6:盐酸维拉佐酮iv晶的制备(06)
将44g1-[4-(5-氰基吲哚-3-基)丁基]-4-(2-氨甲酰-苯并呋喃-5-基)-哌嗪加入到6600ml异丙醇中,加热至回流,回流1小时后趁热过滤,将滤液加热到65℃,滴加含35%氯化氢的异丙醇溶液10.4ml,滴加过程中保持温度为65℃,持续0.2小时,滴加完毕后加热至回流,回流6小时,过滤,在80-100℃,真空度-0.09~-0.1mpa条件下烘干25小时,收料,制得盐酸维拉佐酮iv晶(04)42.0g,纯度是97.89%,收率88.17%。
实施例7:盐酸维拉佐酮iv晶的制备(07)
将4.4kg1-[4-(5-氰基吲哚-3-基)丁基]-4-(2-氨甲酰-苯并呋喃-5-基)-哌嗪加入到308l异丙醇中,加热至回流,回流1小时后趁热过滤,将滤液加热到75℃,滴加含15%氯化氢的异丙醇溶液2.43l,滴加过程中保持温度为75℃,持续1小时,滴加完毕后加热至回流,回流2小时,过滤,在80-100℃,真空度-0.09~-0.1mpa条件下烘干12小时,收料,制得盐酸维拉佐酮iv晶(05)4.40kg,纯度是99.03%,收率92.37%。
实施例8:盐酸维拉佐酮iv晶(07)的晶型数据
盐酸维拉佐酮iv晶(07)的x衍射图谱详见附图1,晶型数据如下表所示。
表1盐酸维拉佐酮iv晶(05)的晶型数据
实施例9:盐酸维拉佐酮iv晶(02)的水分、粒度和残留溶剂
1、盐酸维拉佐酮iv晶(02)的水分,以160928批为例,干燥失重:0.31%。
2、盐酸维拉佐酮iv晶(02)的粒度(见附图2)
d90=58.71μm>50μm。
3、盐酸维拉佐酮iv晶(02)的残留溶剂(见附图4、5)
称取本品样品约0.2g,置顶空瓶中,精密加入二甲基亚砜2ml,密封,振摇使溶解,作为供试品溶液。另取甲醇、乙醇、2-氯丙烷、丙酮、异丙醇、二氯甲烷、四氢呋喃、甲苯、n,n-二甲基甲酰胺适量,精密称定,用二甲基亚砜定量稀释制成对照品溶液。其中,对照品溶液中每1ml含异丙醇约500μg。照残留溶剂测定法(中国药典2015年版四部通则0861第二法)试验。
取对照品溶液顶空注入气相色谱仪中,出峰顺序依次为甲醇、乙醇、2-氯丙烷、丙酮、异丙醇、二氯甲烷、四氢呋喃、甲苯、n,n-二甲基甲酰胺、二甲基亚砜,各成分峰之间的分离度均应符合要求。再取供试品溶液和对照品溶液分别顶空注入气相色谱仪,记录色谱图。要求按外标法以峰面积计算,异丙醇不得过0.5%。
实际盐酸维拉佐酮iv晶(02)残留溶剂以160928批为例,样品异丙醇含量约为1.10%,超过标准限度(0.5%),如下表所示:
表2盐酸维拉佐酮iv晶(02)的残留溶剂
实施例10:盐酸维拉佐酮iv晶(07)的水分、粒度和残留溶剂
1、盐酸维拉佐酮iv晶(07)的水分,以r160907批为例,干燥失重:0.17%。
2、盐酸维拉佐酮iv晶(07)的粒度(见附图3)
d90=42.30μm<50μm。
3、盐酸维拉佐酮iv晶(07)的残留溶剂(见附图6、7)
称取本品样品约0.2g,置顶空瓶中,精密加入二甲基亚砜2ml,密封,振摇使溶解,作为供试品溶液。另取甲醇、乙醇、2-氯丙烷、丙酮、异丙醇、二氯甲烷、四氢呋喃、甲苯、n,n-二甲基甲酰胺适量,精密称定,用二甲基亚砜定量稀释制成对照品溶液。其中,对照品溶液中每1ml含异丙醇约500μg。照残留溶剂测定法(中国药典2015年版四部通则0861第二法)试验。
取对照品溶液顶空注入气相色谱仪中,出峰顺序依次为甲醇、乙醇、2-氯丙烷、丙酮、异丙醇、二氯甲烷、四氢呋喃、甲苯、n,n-二甲基甲酰胺、二甲基亚砜,各成分峰之间的分离度均应符合要求。再取供试品溶液和对照品溶液分别顶空注入气相色谱仪,记录色谱图。要求按外标法以峰面积计算,异丙醇不得过0.5%。
实际盐酸维拉佐酮iv晶(07)残留溶剂以r160907批为例,样品异丙醇含量为0.44%,小于超过标准限度(0.5%),如下表所示:
表3盐酸维拉佐酮iv晶(07)的残留溶剂
对比实施例11:盐酸维拉佐酮iv晶(02)制备的片剂的溶出曲线
盐酸维拉佐酮iv晶(02),批号160928,自制的片剂(自制制剂),批号1611108。对自制制剂1611108进行体外溶出试验,溶出介质:ph4.0缓冲液。
表4盐酸维拉佐酮iv晶(02)的累积溶出度
自制制剂与参比制剂的溶出曲线,见附图8。
可见,由上表和附图8可知,由盐酸维拉佐酮iv晶(02),批号160928,自制的片剂(自制制剂),批号1611108,在ph4.0缓冲液中的体外累积溶出度与原研参比制剂相比,在各时间点均有较大差距,通过附图可见,两者溶出曲线不能拟合,预示着两者体内的溶出性质将有很大差别,进而产生生物利用度和疗效的差异。
实施例12:盐酸维拉佐酮iv晶(07)制备的片剂的溶出曲线
盐酸维拉佐酮iv晶(07),批号r160907,自制的片剂(自制制剂),批号171026。对自制制剂171026进行体外溶出试验,溶出介质:ph4.0缓冲液。
表5盐酸维拉佐酮iv晶(07)的累积溶出度
自制制剂与参比制剂的溶出曲线,见附图9。
可见,由上表和附图9可知,由盐酸维拉佐酮iv晶(07),批号r160907,自制的片剂(自制制剂),批号171026,在ph4.0缓冲液中的体外累积溶出度与原研参比制剂相比,各时间点均差异很小,通过附图可见两者溶出曲线可以拟合,预示着两者在体内的溶出性质可能没有差别,此自制制剂的生物利用度和疗效与原研更为接近。
需要说明的是,上述发明内容及具体实施方式意在证明本发明所提供技术方案的实际应用,不应解释为对本发明保护范围的限定。本领域技术人员在本发明的精神和原理内,当可作各种修改、等同替换、或改进。本发明的保护范围以所附权利要求书为准。
- 还没有人留言评论。精彩留言会获得点赞!