一种制备丝柏油醇的方法与流程
本发明涉及丝柏油醇的制备技术领域,尤其涉及丝柏油醇的产业化及高纯度的制备方法。
背景技术:
丝柏油醇又称扁柏酚或桧木醇(hinokitiol,β-thujaplicin),是从中国台湾扁柏、日本青森丝柏树材中提取的一种具有七环卓酚酮骨架的单萜类的天然化合物,属托酚酮族化合物,具有良好的广谱抗菌性、保湿性和害虫忌避效果,是高安全性的植物成分,可做为抗菌、防虫剂的制备原料。作为台湾扁柏精油的主要成分,它具有较为广泛的生物活性和较强的杀菌能力,对一般细菌,其最小抑菌浓度为10-100ppm,且气味芬芳而效果良好,不仅能杀死空气中细菌、霉菌,亦能防止害虫侵害人体,从而抑制人类病源菌;特别是针对金黄色葡萄球菌(mrsa)有惊人的抑制效果。该mrsa菌常存于人体的皮肤及呼吸道,往往会引起疮疖脓疡等皮肤感染,甚至会导致败血症、腹膜炎、食物中毒等重症疾病。
另外,丝柏油醇在美容美白方面的应用,对酪氨酸酶双酚酶具有很强的抑制作用,以多巴为底物,导致酶活力下降50%的丝柏油醇浓度(ic50)为0.3μmol/l。酪氨酸酶是黑色素形成的关键酶,在人体内能催化轻氨酸、多巴通过多条途径转化成优黑素、褐色素等,从而引起雀斑、黄褐斑、妊娠斑的生成。体外实验表明,丝柏油醇抑制酪氨酸酶活力而不致导致酶的永久失活,对黑色素细胞中酪氨酸酶的调控机制与其结构有密切关系。酪氨酸酶是一种含酮的氧化还原酶,而丝柏油醇环庚三烯上2位的羟基和1位的酮结构恰能与酪氨酸酶活性中心铜原子相结合,形成紧密的络合物,从而导致酪氨酸酶催化活性的下降,这是丝柏油醇抑制酪氨酸酶活力的主要方式。同时,丝柏油醇上的羟基和酮结构亦能与酪氯酸酶的底物络合物(es)结合,形成三元络合物进而调控黑色素的形成。
近年来,围绕丝柏油醇具广谱抗菌抗霉、抑制病毒活性、抑制酪氨酸酶及植物生成乙烯等作用以及具有赋予细胞活性的特点,将其应用在医药开发、卫生用品、化妆品、养发育毛、食品保鲜等诸多产品的开发上备受业界关注。
传统的丝柏油醇的制备,以甲氧基环庚(间)三烯,经异丙基环庚三烯酮、胺基异丙基环庚三烯酮过程;香芹酮环氧化-缩醛化;异丙基环己酮(或烯)氰醇化,以及利用溴环庚三烯酚酮与有机锡化合物作用等方法,其合成步骤多,且原料难以得到,几乎无法施于工业生产,故缺乏产业化生产的实用性。
近年来,丝柏油醇制备围绕产业化生产着眼如下的发展方向实施,以环戊二烯为原料与格林尼亚试剂二乙基溴化镁和异丙基甲苯磺酸盐反应,或在碱性条件下与丙酮反应,或与脂肪低级醇或乙烯的气相催化反应,或与碱金属制得环戊二烯金属化合物经卤化烷基反应等烷基化得到烷基环戊二烯,制备丝柏油醇等系列方法。由于这些方法过程中使用的格林尼亚、氢化二烷基铝、烷基锂等试剂价格昂贵,且由低温控制逐渐升温的过程中,对气相条件的设备工艺要求严苛;由于易生成多取代烷基体,导致单烷基体收率低,而且产物的平衡组成基本为1-异丙基环戊二烯和2-异丙基环戊二烯的等量异构体混合物,它不适合制备高纯度的丝柏油醇。再有,原料环戊二烯属于含能化合物(火箭推进剂),且沸点较低,室温下极不稳定,容易形成二聚体,并在形成二聚体的过程中放出大量的热而造成爆炸。由此导致丝柏油醇的制备不能进行大规模的产业化与应用。
因此,现有的丝柏油醇的制备技术亟需在以下方面进行改进:
(1)找一种替代具含能特性的环戊二烯作为制备的初始原料,能够最大程度地降低原料保存成本及提高生产安全性;
(2)避免烷基锂/格氏试剂等活性较强的化合物参与相关反应步骤,从而消除丝柏油醇规模化生产过程中的安全隐患;
(3)避免在制备过程中出现同分异构体,从而提高产业化制备丝柏油醇的产率和纯度。
技术实现要素:
本发明所要解决的技术问题是提供一种制备丝柏油醇的方法,能够最大程度地提高丝柏油醇产业化生产的安全性及降低物料的保存成本。
为了解决上述技术问题,本发明提供了一种制备丝柏油醇的方法,包括:
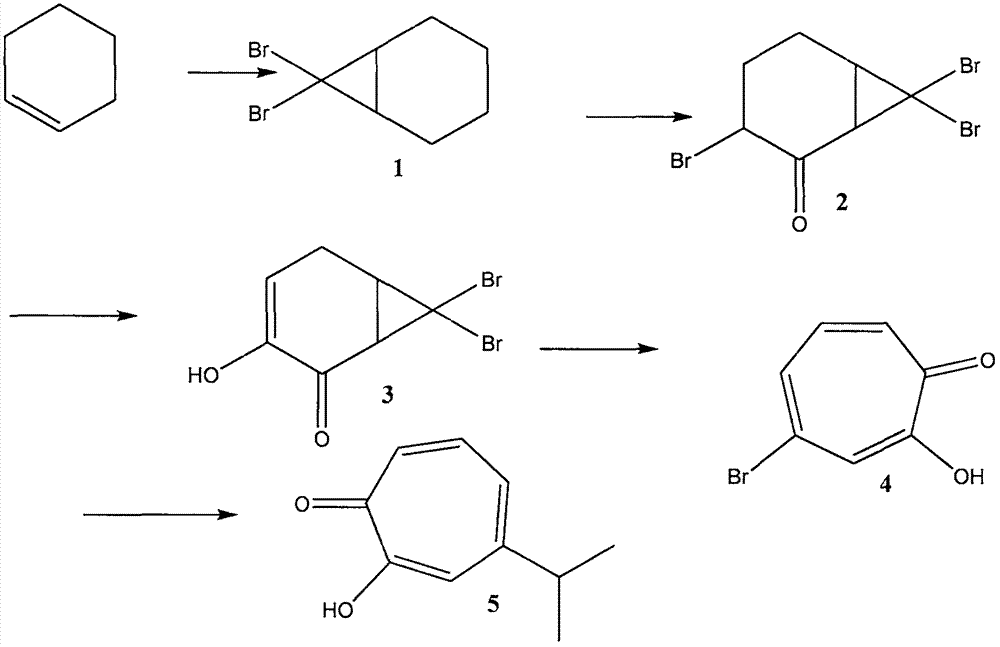
将起始原料化合物环己烯与三溴甲烷进行加成反应得到称为7,7-二溴双环[4.1.0]庚烷的第一化合物;该第一化合物经过氧化反应得到称为3,7,7-三溴双环[4.1.0]庚-2-酮的第二化合物;该第二化合物经过取代反应得到称为7,7-二溴-3-羟基双环[4.1.0]庚-3-烯-2-酮的第三化合物;该第三化合物经过扩环反应得到称为4-溴-2-羟基环庚-2,4,6-三烯-1-酮的第四化合物;该第四化合物经过烷基化反应得到称为丝柏油醇的第五化合物。
优选地,第一化合物的制备具体包括如下步骤:
将环己烯与三溴甲烷混合并在三乙基苯基氯化铵和无机碱的催化下进行加成反应,反应温度为-20℃~30℃,反应时间为0.5~30小时;其中,环己烯与三溴甲烷的体积比为1∶0.5~1∶100,环己烯与三乙基苯基氯化铵的体积比为1000∶1~10∶1;
加成反应完毕用盐酸淬灭,该盐酸的浓度为1m~10m;然后用二氯甲烷溶液萃取分离,从该二氯甲烷溶液中收集第一化合物。
优选地,环己烯与三溴甲烷的体积比为进一步选为1∶2~1∶10,环己烯与三乙基苯基氯化铵的体积比进一步选为100∶1~50∶1;无机碱包括氢氧化钠、氢氧化钾中的任意一种。
优选地,第二化合物的制备具体包括如下步骤:
将第一化合物1与三氧化铬在乙醚的助剂下进行氧化反应,反应温度为-20℃~40℃,反应时间为0.5~8小时;其中,第一化合物与所述三氧化铬的质量比为1∶30~1∶2,第一化合物与所述乙醚的重量体积比为0.2g/ml~0.01g/ml;
所述氧化反应完毕将反应物倾入水中,用乙醚萃取分离收集到第二化合物。
优选地,第一化合物与三氧化铬的质量比进一步选为:1∶5~1∶3;第一化合物与乙醚的重量体积比选一步选为0.1g/ml~0.04g/ml。
优选地,第三化合物的制备具体包括如下步骤:
将第二化合物与二甲基亚砜即dmso有机溶剂在有机碱的催化下进行羟基取代反应,反应温度为60℃~150℃,反应时间为1~10小时;其中,第二化合物与dmso的重量体积比为0.5g/ml到0.001g/ml;第二化合物与有机碱的摩尔比为1∶0.2~1∶30;
羟基取代反应完毕用盐酸淬灭,该盐酸的浓度为0.5m~6m;然后用二氯甲烷溶液萃取分离收集到第三化合物。
优选地,第二化合物与dmso的重量体积比进一步选为0.1g/ml~0.01g/ml;第二化合物与有机碱的摩尔比进一步选为1∶1~1∶10;有机碱包括吡啶、三乙胺、醇钠中的一种或多种。
优选地,第四化合物的制备具体包括如下步骤:
在惰性气体保护下,将第三化合物和1,8-二氮杂双环(5.4.0)十一碳-7-烯在四氢呋喃溶液中进行扩环反应,反应温度为-70℃~0℃,反应时间为0.5~5小时;其中,第三化合物与1,8-二氮杂双环(5.4.0)十一碳-7-烯之间的摩尔比为1∶0.0001~1∶0.2;第三化合物与四氢呋喃的重量体积比为0.4g/ml~0.002g/ml;
扩环反应完毕,用浓度为0.1m~10m的无机碱溶液进行淬灭;淬灭后用盐酸调节ph至4~5;然后用二氯甲烷萃取分离收集到第四化合物。
优选地,惰性气体包括氮气、氩气中的任意一种;第三化合物与1,8-二氮杂双环(5.4.0)十一碳-7-烯之间的摩尔比进一步选为1∶0.0005~1∶0.001;第三化合物与四氢呋喃的重量体积比进一步选为0.08g/ml~0.01g/ml;无机碱包括氢氧化钠、氢氧化钾中的任意一种。
优选地,第五化合物的制备具体包括如下步骤:
将第四化合物和三甲基丙烯基锡混合在1,4-二氧六环中,并在双三苯基磷二氯化钯催化剂的催化下进行烷基化反应,反应温度为70℃~180℃,反应时间为0.5~5小时;其中,第四化合物与三甲基丙烯基锡之间的摩尔比为1∶0.5~1∶20;第四化合物与1,4-二氧六环的重量体积比为0.2g/ml~0.001g/ml;
烷基化反应完毕,过滤除去反应物中的固体,蒸馏除去溶剂,将残余物溶解在乙醇中,在0.5-10%的钯碳催化下进行氢化还原反应,其中氢气的压力为0.1mpa~2mpa,该氢化还原反应的时间为6~48小时,残余物与乙醇的重量体积比为0.3g/ml~0.001g/ml,该氢化还原反应的温度为0℃~150℃;
氢化还原反应完毕,过滤除去双三苯基磷二氯化钯催化剂,蒸馏,除去乙醇,得到第五化合物。
优选地,第四化合物与三甲基丙烯基锡之间的摩尔比进一步选为1∶0.8~1∶2;第四化合物与1,4-二氧六环的重量体积比进一步优选为0.07g/ml~0.01g/ml;在氢化还原反应中,优选在5%的钯碳催化下进行,氢气的压力进一步选为0.1mpa~0.5mpa;并且,该氢化还原反应的时间进一步选为18~26小时;残余物与乙醇的重量体积比进一步选为0.07g/ml~0.01g/ml;该氢化还原反应的温度进一步优选为20℃~50℃。
本发明提供的产业化制备丝柏油醇的方法,通过使用环己烯作为初始原料,无论在原料的保存成本上,还是安全性方面都有了很大程度的提高,根本性地解决了产业化生产的安全性问题;由于本发明的方法通过先制备出七元环结构再引入异丙基的过程,避免同分异构体的出现,从而确保了规模化生产的效率;再有,本发明的方法避开了烷基锂/格氏试剂等活性较强的化合物参与相关反应的步骤,从而消除了丝柏油醇规模化生产过程中的安全隐患。
附图说明
图1为本发明制备丝柏油醇的方法的反应通式表达;
图2为本发明制备丝柏油醇的方法中化合物1的制备其反应通式表达;
图3为本发明制备丝柏油醇的方法中化合物2的制备其反应通式表达;
图4为本发明制备丝柏油醇的方法中化合物3的制备其反应通式表达;
图5为本发明制备丝柏油醇的方法中化合物4的制备其反应通式表达;
图6为本发明制备丝柏油醇的方法中目标化合物5的制备其反应通式表达。
具体实施方式
以下结合优选实施例对本发明的技术方案进行详细地阐述。
本发明提供的一种制备丝柏油醇的方法,包括:将起始原料化合物环己烯与三溴甲烷进行加成反应得到化合物1(即7,7-二溴双环[4.1.0]庚烷);该化合物1经过氧化反应得到化合物2(即3,7,7-三溴双环[4.1.0]庚-2-酮);该化合物2经过取代反应得到化合物3(即7,7-二溴-3-羟基双环[4.1.0]庚-3-烯-2-酮);该化合物3经过扩环反应得到化合物4(即4-溴-2-羟基环庚-2,4,6-三烯-1-酮);最后该化合物4经过烷基化反应得到目标化合物5(即丝柏油醇)。
该制备的反应通式如图1中所示。该图中,标号1、2、3、4、5分别表示化合物1、化合物2、化合物3、化合物4及目标化合物5。
在本发明的制备丝柏油醇的方法中,化合物1的制备包括如下步骤:
将环己烯与三溴甲烷混合并在三乙基苯基氯化铵和无机碱的催化下进行加成反应,反应温度为-20℃~30℃,反应时间为0.5~30小时;其中,环己烯与三溴甲烷的体积比为1∶0.5~1∶100,优选为1∶2~1∶10,环己烯与三乙基苯基氯化铵的体积比为1000∶1~10∶1,优选为100∶1~50∶1;无机碱包括氢氧化钠、氢氧化钾中的任意一种;
反应完毕用盐酸淬灭,盐酸浓度为1m~10m;然后用二氯甲烷萃取分离,从该二氯甲烷溶液中收集化合物1,收率为73%。
该制备的反应通式如图2中所示。该图中,标号1表示化合物1。
实施例1化合物1的合成制备
将10毫升环己烯溶解在50毫升三溴甲烷中,加入三乙基苯基氯化铵(相转移催化剂)0.1克,将上述溶液冷却至0℃;快速搅拌后滴加24克50%的氢氧化钠水溶液(亲核反应催化剂),于0℃下搅拌2小时,再于室温下继续搅拌22小时;然后冷却至0℃,滴加6m的盐酸30毫升;分离出有机相,水相中残留的化合物1用二氯甲烷萃取;通过无水硫酸镁干燥有机相,除去干燥剂、溶剂;通过减压蒸馏得到无色的液体化合物1,收率达73%。
化合物1的结构通过氢核磁共振谱(hnmr)和质谱(esi-ms)进行表征,相关数据如下:
1hnmr(cdcl3,300mhz),δ:2.38(t,2h),1.32-1.78(m,8h);esi-ms:251(m+)。
在本发明的制备丝柏油醇的方法中,化合物2的制备包括如下步骤:
将化合物1与三氧化铬在乙醚的助剂下进行氧化反应,反应温度为-20℃~40℃,反应时间为0.5-8小时;其中,化合物1与三氧化铬的质量比为1∶30~1∶2,优选为:1∶5~1∶3;化合物1与乙醚的重量体积比为0.2g/ml~0.01g/ml,优选为0.1g/ml~0.04g/ml;
反应完毕将反应物倾入水中,用乙醚萃取分离收集到化合物2,收率为90%。
该制备的反应通式如图3所示。该图中,标号1、2分别表示化合物1及化合物2。
实施例2化合物2的合成制备
0℃下将7.6克化合物1加入到200毫升三氧化铬(30克)的乙醚溶液中,并于25-30℃下搅拌1小时,将反应物倾入到1升水中,分离有机相,水相中残留的化合物2用乙醚萃取;混合分离出的有机相和萃取液,并用碳酸钠水溶液、饱和食盐水洗涤,以去除有机相中所残留的杂质;用无水硫酸镁干燥后,蒸馏除去乙醚,得到化合物2(6.8克,产率90%)。
化合物2的结构通过氢核磁共振谱和质谱进行表征,相关数据如下:
1hnmr(cdcl3,300mhz),δ:4.32(t,1h),4.08(d,1h),3.91(m,1h),2.42(m,2h),1.96(m,2h);esi-ms:343(m+)。
在本发明的制备丝柏油醇的方法中,化合物3的制备包括如下步骤:
将化合物2与二甲基亚砜(dmso,dimethylsulfoxide)有机溶剂在有机碱的催化下进行羟基取代反应,反应温度为60℃~150℃,反应时间为1~10小时;其中,化合物2与dmso的重量体积比为0.5g/ml到0.001g/ml,优选为0.1g/ml~0.01g/ml;化合物2与有机碱的摩尔比为1∶0.2~1∶30,优选为1∶1~1∶10;所用有机碱包括吡啶、三乙胺、醇钠中的一种或多种;
反应完毕用盐酸进行淬灭,盐酸的浓度为0.5m~6m;然后用二氯甲烷萃取分离收集到化合物3,收率为87%。
该制备的反应通式如图4所示。该图中,标号2、3分别表示化合物2及化合物3。
实施例3化合物3的合成制备
将9克吡啶,13克化合物2溶解在300毫升dmso中,加热至90-95℃,并保持此温度4小时;再冷却至室温,加入200毫升二氯甲烷和2m的盐酸300毫升;搅拌均匀后,分出有机相;水相中残留的化合物3用二氯甲烷萃取;混合分离出的有机相和萃取液,用水洗、干燥,除去二氯甲烷得到化合物3(11g)产率达87%。
化合物3的结构通过氢核磁共振谱和质谱进行表征,相关数据如下:
1hnmr(cdcl3,300mhz),δ:5.98(t,1h),3.18(d,1h),2.92(m,1h),1.96(m,2h);esi-ms:279(m+)。
在本发明的制备丝柏油醇的方法中,化合物4的制备包括如下步骤:
在惰性气体保护下,将化合物3和1,8-二氮杂双环(5.4.0)十一碳-7-烯在四氢呋喃溶液中进行扩环反应,反应温度为-70℃~0℃,反应时间为0.5~5小时;其中,惰性气体包括氮气、氩气中的任意一种;化合物3与1,8-二氮杂双环(5.4.0)十一碳-7-烯之间的摩尔比为1∶0.0001~1∶0.2,优选为1∶0.0005~1∶0.001;化合物3与四氢呋喃的重量体积比为0.4g/ml~0.002g/ml,优选为0.08g/ml~0.01g/ml;
反应完毕用浓度为0.1m~10m的无机碱溶液进行淬灭,无机碱包括氢氧化钠、氢氧化钾中的任意一种;淬灭后用盐酸调节ph至4~5;然后用二氯甲烷萃取分离收集到化合物4,收率为62%。
该制备的反应通式如图5所示。该图中,标号3、4分别表示化合物3及化合物4。
实施例4化合物4的合成制备
在氮气保护和温度-40℃条件下将1.4毫克的1,8-二氮杂双环(5.4.0)十一碳-7-烯(dbu)加入到1.45克化合物3的50毫升四氢呋喃溶液中,并于低温下搅拌2小时,再补加0.7毫克的dbu,缓慢将温度升至0℃;将20毫升2m的氢氧化钠溶液加入到上述反应物中,搅拌5分钟;用2m的盐酸将其酸化,并用二氯甲烷萃取水相中残余的化合物4;用无水硫酸镁干燥萃取液,除去干燥剂、二氯甲烷得到化合物4(0.6克),产率达62%。
化合物4的结构通过氢核磁共振谱和质谱进行表征,相关数据如下:
1hnmr(cdcl3,300mhz),δ:7.65(s,1h),7.52(d,1h),7.10(d,1h),6.73(d,1h);esi-ms:199(m+)。
在本发明的制备丝柏油醇的方法中,化合物5的制备包括如下步骤:
将化合物4和三甲基丙烯基锡混合在1,4-二氧六环中,并在双三苯基磷二氯化钯催化下进行烷基化反应,反应温度为70℃~180℃,反应时间为0.5~5小时;其中,化合物4与三甲基丙烯基锡之间的摩尔比为1∶0.5~1∶20,优选为1∶0.8~1∶2;化合物4与1,4-二氧六环的重量体积比为0.2g/ml~0.001g/ml,优选为0.07g/ml~0.01g/ml;
反应完毕,过滤除去反应物中的固体,蒸馏除去溶剂,将残余物溶解在乙醇中,在钯碳催化下进行氢化还原反应;
该氢化还原反应完毕,过滤除去催化剂,蒸馏,除去乙醇,得到化合物5,收率为53%。
氢化还原过程中,加入0.5-10%的钯碳,优选为加入5%,氢气的压力为0.1mpa~2mpa,优选为0.1mpa~0.5mpa;氢化时间为6~48小时,优选为18~26小时;残余物与乙醇的重量体积比为0.3g/ml~0.001g/ml,优选为0.07g/ml~0.01g/ml;氢化温度为0℃~150℃,优选为20℃~50℃。
该制备的反应通式如图6所示。该图中,标号4、5分别表示化合物4及化合物5。
实施例5化合物5的合成制备
将2克化合物4、三甲基丙烯基锡3克和催化剂双三苯基磷二氯化钯0.3克混合加入到100毫升1,4-二氧六环中,回流1个小时,进行烷基化反应;过滤除去反应过程中所析出的固体,干燥,除去1,4-二氧六环;将去除溶剂后的残留物加入到100毫升乙醇中,再加入5%的钯碳,保持氢气压力1个大气压进行氢化还原反应;室温下反应24小时后,过滤,除去固体催化剂和乙醇得到化合物5(0.94克),产达率53%。
化合物5的结构通过氢核磁共振谱和质谱进行表征,相关数据如下:
1hnmr(cdcl3,300mhz),δ:7.65(s,1h),7.52(d,1h),7.08(d,1h),6.70(d,1h);esi-ms:164(m+)。
综上,本发明通过使用环己烯作为初始原料,使得无论在原料的保存成本上,还是安全性方面都有了很大程度的提高,因而从根本性地解决了丝柏油醇产业化规模化生产的安全性问题;另外,由于本发明的方法通过先制备出七元环结构再引入异丙基的过程,避免同分异构体的出现,从而确保了规模化生产的产率和较高的纯度;再有,本发明的方法避开了烷基锂/格氏试剂等活性较强的化合物参与相关反应的步骤,从而消除了丝柏油醇规模化生产过程中的安全隐患;。故可见本发明的技术方案取得的有益效果的非常显著。
- 还没有人留言评论。精彩留言会获得点赞!