聚氨酯的制造方法以及环氧羧酸酯组合物、聚氨酯和聚氨酯树脂组合物与流程
本发明涉及聚氨酯的制造方法以及环氧羧酸酯组合物、聚氨酯和聚氨酯树脂组合物。
背景技术:
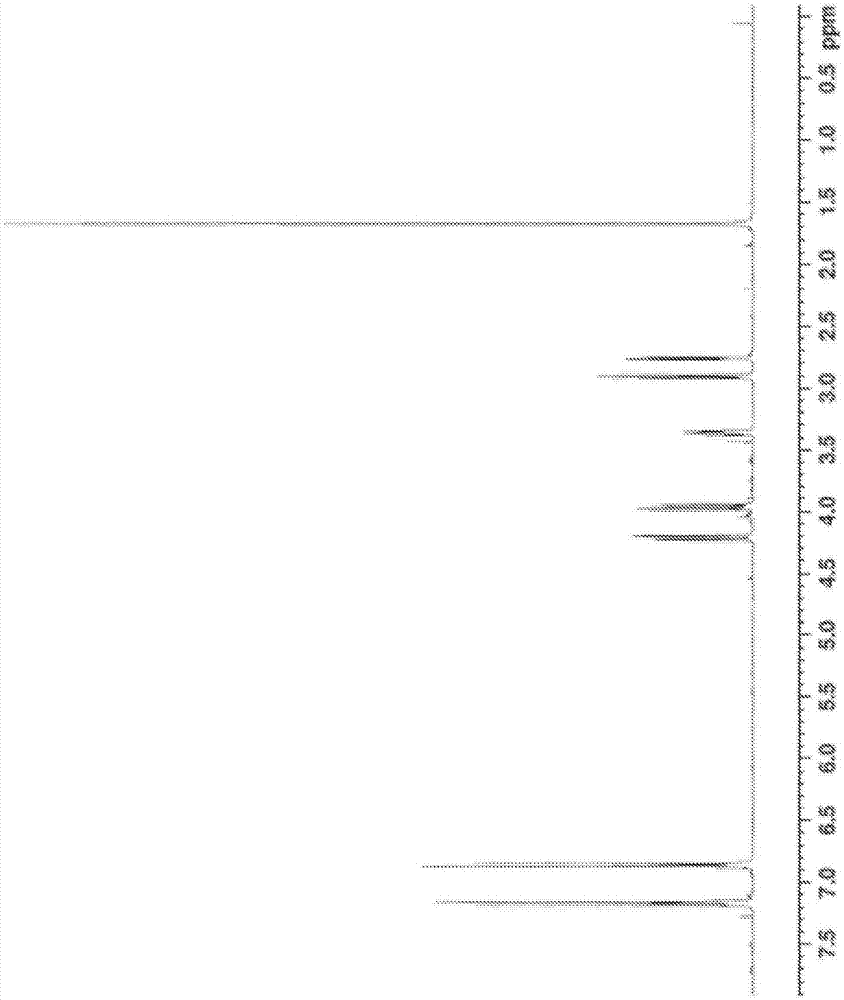
:以往,聚氨酯化合物使用于各种用途。例如,在电子材料领域中,可举出阻焊剂、磁盘用材料、膜涂布材料等用途。在下述专利文献1中公开了对柔性基板的阻焊剂用途的聚氨酯化合物。此外,在下述专利文献2中公开了光盘用树脂组合物用途的聚氨酯化合物。此外,在下述专利文献3中公开了膜涂布材料用途的聚氨酯化合物。在上述专利文献1中记载了,在合成作为聚氨酯化合物的原料的环氧羧酸酯(包含环氧丙烯酸酯)化合物时优选使用不具有羟基的环氧化合物,但一般而言丙烯酸系固化膜的固化收缩剧烈,在整面涂布时基材的翘曲成为问题。然而,专利文献1中,对柔性基板实施部分涂布,并未评价在柔性基板上整面涂布时的翘曲。此外,也未进行耐溶剂性的评价。此外,在上述专利文献2中,使用分子中具有2个以上环氧基的环氧化合物来合成环氧羧酸酯化合物,聚氨酯化合物的原料中含有包含3个以上羟基的环氧羧酸酯化合物的可能性高。因此存在下述问题:在合成聚氨酯化合物时,原料粘度变高而操作变得困难,而且聚氨酯变为多支链结构而聚合控制也变难。此外,在专利文献3中,使用包含2个以上羟基的环氧化合物,氨基甲酸酯(甲基)丙烯酸酯化合物的原料使用包含3个以上羟基的(甲基)丙烯酸酯化合物。因此,产生与上述专利文献2同样的问题。此外,关于以上述以往的环氧(甲基)丙烯酸酯作为原料的聚氨酯化合物,来源于原料的氯等卤素原子的含有率高,在例如电子材料领域中使用的情况下,具有使作为导电材料的铜等金属发生迁移等的可能性。现有技术文献专利文献专利文献1:日本专利第5638866号公报专利文献2:日本专利第4750789号公报专利文献3:日本专利第5781406号公报技术实现要素:发明所要解决的课题本发明的目的是提供操作容易、聚合控制易于进行、卤素的含有率低、固化后的与基板的密合性、耐固化收缩性、耐溶剂性优异的聚氨酯的制造方法以及环氧羧酸酯组合物、聚氨酯和聚氨酯树脂组合物。用于解决课题的方法为了达到上述目的,本发明包含以下实施方式。[1]一种聚氨酯的制造方法,其包含下述第一工序和第二工序,第一工序是使环氧化合物(a)、不饱和脂肪族一元羧酸(b)、和芳香族一元羧酸(c)进行反应而获得环氧羧酸酯化合物(x)的工序,所述环氧化合物(a)的卤素原子的含量为10质量ppm以下,并且其分子中具有2个环氧基且不具有羟基,所述不饱和脂肪族一元羧酸(b)的分子中具有烯属不饱和基且不具有芳香环,所述芳香族一元羧酸(c)的分子中不具有烯属不饱和基;第二工序是使所述第一工序中获得的环氧羧酸酯化合物(x)与二异氰酸酯化合物(y)进行反应的工序。[2]根据[1]所述的聚氨酯的制造方法,在上述第一工序中,在相对于上述环氧化合物(a)的环氧基1当量、分子中具有烯属不饱和基且不具有芳香环的不饱和脂肪族一元羧酸(b)与分子中不具有烯属不饱和基的芳香族一元羧酸(c)的羧基的合计成为0.9~1.2当量的条件下反应,分子中不具有烯属不饱和基的芳香族一元羧酸(c)相对于分子中具有烯属不饱和基且不具有芳香环的不饱和脂肪族一元羧酸(b)与分子中不具有烯属不饱和基的芳香族一元羧酸(c)的合计的比例为20~80摩尔%。[3]根据[1]或[2]所述的聚氨酯的制造方法,在上述第二工序中,二异氰酸酯化合物(y)的异氰酸酯基:环氧羧酸酯化合物(x)的羟基为0.5~1.5:1。[4]根据[1]~[3]中任一项所述的聚氨酯的制造方法,上述环氧化合物(a)是卤素原子的含量为10质量ppm以下的芳香族二缩水甘油基醚。[5]根据[4]所述的聚氨酯的制造方法,上述芳香族二缩水甘油基醚为双酚a型二缩水甘油基醚。[6]根据[1]~[5]中任一项所述的聚氨酯的制造方法,上述分子中具有烯属不饱和基且不具有芳香环的不饱和脂肪族一元羧酸(b)为由(甲基)丙烯酸、巴豆酸、(甲基)丙烯酸与ε-己内酯的反应生成物、下述半酯所组成的组中的任一种,所述半酯是分子中不具有芳香环的饱和或不饱和二元酸酐与1分子中具有1个羟基的(甲基)丙烯酸酯衍生物的等摩尔反应物。[7]根据[1]~[6]中任一项所述的聚氨酯的制造方法,上述分子中不具有烯属不饱和基的芳香族一元羧酸(c)是具有碳原子数为6~30的芳香环骨架的一元羧酸。[8]根据[1]~[7]中任一项所述的聚氨酯的制造方法,上述二异氰酸酯化合物(y)包含除异氰酸酯基(-nco基)中的碳原子以外的碳原子的数目为6~30的脂环式化合物。[9]一种环氧羧酸酯组合物,其包含下述式(1)所示的第一环氧羧酸酯化合物和下述式(1)所示的第二环氧羧酸酯化合物,(式中,a为来源于二缩水甘油基醚的有机残基,且不具有羟基。r1、r2各自独立地为可以具有取代基的碳原子数为6~30的芳香环或式(2)所示的有机基团,上述第一环氧羧酸酯化合物的r1、r2中的至少一个为可以具有取代基的碳原子数为6~30的芳香环,上述第二环氧羧酸酯化合物的r1、r2中的至少一个为式(2)所示的有机基团,第一环氧羧酸酯化合物与第二环氧羧酸酯化合物不同。)h2c=cr3-(2)(式中,r3表示氢原子或甲基。)[10]一种聚氨酯,其包含下述式(1)所示的第一环氧羧酸酯化合物与二异氰酸酯化合物(y)的第一氨基甲酸酯键单元、和下述式(1)所示的第二环氧羧酸酯化合物与二异氰酸酯化合物(y)的第二氨基甲酸酯键单元。(式中,a为来源于二缩水甘油基醚的有机残基,且不具有羟基。r1、r2各自独立地为可以具有取代基的碳原子数为6~30的芳香环或式(2)所示的有机基团,上述第一环氧羧酸酯化合物的r1、r2中的至少一个为可以具有取代基的碳原子数为6~30的芳香环,上述第二环氧羧酸酯化合物的r1、r2中的至少一个为式(2)所示的有机基团,第一环氧羧酸酯化合物与第二环氧羧酸酯化合物不同。)h2c=cr3-(2)(式中,r3表示氢原子或甲基。)[11]一种聚氨酯树脂组合物,其包含上述[10]所述的聚氨酯、和光引发剂。发明效果根据本发明,能够制造操作容易、聚合控制易于进行、卤素的含有率低、固化后的与基板的密合性、耐固化收缩性、耐溶剂性优异的聚氨酯。附图说明图1是表示通过合成例1获得的双酚a型二缩水甘油基醚(badg)的1h-nmr光谱的图。图2是表示通过合成例2获得的双酚a型二缩水甘油基醚(badg)丙烯酸酯的1h-nmr光谱的图。图3是表示通过合成例6获得的双酚a型二缩水甘油基醚(badg)丙烯酸酯的1h-nmr光谱的图。具体实施方式以下,对用于实施本发明的方式(以下,称为实施方式)进行说明。实施方式涉及的聚氨酯的制造方法,其特征在于,包含下述第一工序和第二工序,第一工序是使卤素原子的含量为10质量ppm以下、并且分子中具有2个环氧基且不具有羟基的环氧化合物(a)(以下,有时简称为环氧化合物(a))、分子中具有烯属不饱和基且不具有芳香环的不饱和脂肪族一元羧酸(b)(以下,有时简称为不饱和脂肪族一元羧酸(b))、和分子中不具有烯属不饱和基的芳香族一元羧酸(c)(以下,有时简称为芳香族一元羧酸(c))进行反应而获得环氧羧酸酯化合物(x);第二工序是使上述第一工序中获得的环氧羧酸酯化合物(x)与二异氰酸酯化合物(y)进行反应。这里,上述卤素原子主要是氯原子和溴原子。因此,所谓卤素原子的含量为10质量ppm以下,是指相对于上述环氧化合物(a)总量的总氯原子浓度和总溴原子浓度的合计为10质量ppm以下。通过使作为原料的环氧化合物(a)的卤素原子的含量为10质量ppm以下,能够使合成的聚氨酯的卤素原子的含量为50质量ppm以下,在电子材料领域中使用的情况下可以大幅抑制作为导电材料的铜等金属所产生的迁移等。在本说明书中,卤素原子的含量是依照jisk7243-3测定的值。此外,作为环氧羧酸酯化合物(x)的原料的环氧化合物(a)由于分子中不具有羟基,氢键少,因此粘度低(5~6pa·s左右),操作容易。本实施方式中使用的环氧羧酸酯化合物(x)的粘度在25℃下为400~700pa·s的范围。后述的合成例中,粘度使用antonpaar社制旋转流变仪mcr301在5rpm的条件下测定。<环氧羧酸酯化合物(x)的合成:第一工序>作为合成环氧羧酸酯化合物(x)所使用的分子中具有2个环氧基且不具有羟基的环氧化合物(a),没有特别限制,可举出例如,双酚-a二缩水甘油基醚、双酚-f二缩水甘油基醚、双酚-s二缩水甘油基醚、双酚芴二缩水甘油基醚等双酚系二缩水甘油基醚类、联苯酚二缩水甘油基醚、四甲基联苯酚缩水甘油基醚等联苯系缩水甘油基醚类等芳香族系二缩水甘油基醚化合物;己二醇二缩水甘油基醚等烷基二醇二缩水甘油基醚类、聚乙二醇二缩水甘油基醚等亚烷基二醇二缩水甘油基醚类、环己二醇二缩水甘油基醚、氢化双酚-a二缩水甘油基醚等环烷基二醇二缩水甘油基醚类等饱和烃系二缩水甘油基醚化合物;3,4-环氧环己烯基甲基-3’,4’-环氧环己烯甲酸酯(ダイセル化学(株)制セロキサイド2021)、1,2,8,9-二环氧苧烯(ダイセル化学(株)制セロキサイド3000)等所谓二官能脂环式环氧化合物。此外,环氧化合物(a)除了不具有羟基以外,还不具有羧基、巯基等具有与异氰酸酯基容易反应的活性氢的官能团,这在防止后述的第二工序中的聚氨酯的凝胶化方面是优选的。其中,从最终获得的聚氨酯与基材的密合性的观点考虑,优选为芳香族系二缩水甘油基醚化合物,双酚a二缩水甘油基醚是更适合的。在使用聚对苯二甲酸乙二醇酯(pet)、聚萘二甲酸乙二醇酯(pen)、芳香族聚酰亚胺(pi)等分子中具有芳香环的基材的情况下,如果使用分子中具有芳香环的芳香族系二缩水甘油基醚化合物,则通过最终获得的聚氨酯的芳香环与构成基材的芳香环的π相互作用而两者间的密合性提高。这些环氧化合物(a)可以通过例如日本专利第5800709号公报所记载的方法,使用过氧化氢作为氧化剂,将分子中具有2个碳-碳双键的有机化合物的碳-碳双键进行环氧化来制造。根据该方法,由于不使用表卤代醇作为原料,因此能够获得分子的骨架实质上不含卤素原子的环氧化合物(a)。因此,即使没有高度的洗涤处理也可以获得卤素原子的含量小的环氧化合物(a)。环氧化合物(a)中包含的卤素原子的含量如上所述优选为10质量ppm以下,更优选为8质量ppm以下。如果环氧化合物(a)中包含的卤素原子的含量为10质量ppm以下,则能够使最终获得的聚氨酯中包含的卤素原子的含量为50质量ppm以下,也可以更优选为40质量ppm以下,进一步优选为30质量ppm以下。合成环氧羧酸酯化合物(x)所使用的不饱和脂肪族一元羧酸(b)是向本实施方式中制造的聚氨酯中导入烯属不饱和基,并且将上述环氧化合物(a)转变为能够与异氰酸酯基反应的二醇化合物的不饱和脂肪族一元羧酸,该不饱和脂肪族一元羧酸(b)的烯属不饱和基数优选为1个~4个。作为这样的不饱和脂肪族一元羧酸(b),可举出例如,(甲基)丙烯酸类、巴豆酸等。在本说明书中,“(甲基)丙烯酸”是指丙烯酸和甲基丙烯酸,“(甲基)丙烯酸酯”是指丙烯酸酯和甲基丙烯酸酯,“(甲基)丙烯酰基”是指丙烯酰基和甲基丙烯酰基。作为上述(甲基)丙烯酸类,可举出例如,(甲基)丙烯酸、(甲基)丙烯酸与ε-己内酯的反应生成物、作为分子中不具有芳香环的饱和或不饱和二元酸酐与1分子中具有1个羟基的(甲基)丙烯酸酯衍生物的等摩尔反应物的半酯类等。其中,从容易获得考虑,(甲基)丙烯酸是适合的。在使用了(甲基)丙烯酸的情况下,环氧羧酸酯化合物(x)为环氧(甲基)丙烯酸酯。环氧羧酸酯化合物(x)的合成所使用的芳香族一元羧酸(c)是与上述不饱和脂肪族一元羧酸(b)一起将上述环氧化合物(a)转变为能够与异氰酸酯基反应的二醇化合物的芳香族一元羧酸,其有助于最终获得的聚氨酯的固化收缩的降低。通过将不饱和脂肪族一元羧酸(b)的一部分置换成芳香族一元羧酸(c),从而导入到最终获得的聚氨酯的烯属不饱和基的量降低,因此固化收缩被抑制。此外,由于导入到最终获得的聚氨酯中的芳香环增加,因此如果使用分子中具有芳香环的基材,则通过与该芳香环的π相互作用,聚氨酯与基材的密合性提高。作为芳香族一元羧酸(c),优选为具有碳原子数为6~30的芳香环骨架的、且不含烯属不饱和键的一元羧酸。即,优选为可以具有不含烯属不饱和基的取代基、具体为碳原子数为1~6的烷基、碳原子数1~6的烷氧基等作为取代基的、具有苯环或萘骨架、蒽骨架等稠环等作为芳香环骨架的一元羧酸。可举出例如,苯甲酸、甲基苯甲酸(toluicacid)、叔丁基苯甲酸、均三甲苯酸(mesitylenicacid,二甲基苯甲酸)等烷基取代苯羧酸类、茴香酸、二甲基茴香酸、乙氧基苯甲酸、三甲氧基苯甲酸等烷氧基取代苯羧酸类、苯氧基苯甲酸、苄氧基苯甲酸、或萘甲酸、甲氧基萘甲酸、蒽酮酸等稠环羧酸类等。作为取代基,可举出碳原子数为1~6的烷基、碳原子数1~6的烷氧基等。使上述环氧化合物(a)、不饱和脂肪族一元羧酸(b)、和芳香族一元羧酸(c)进行反应而获得环氧羧酸酯化合物(x)的方法(羧酸酯化反应:第一工序)如下所述。优选为在相对于上述环氧化合物(a)的环氧基1当量、不饱和脂肪族一元羧酸(b)与芳香族一元羧酸(c)的羧基的合计成为0.9~1.2当量的条件下反应。如果为该范围,则能够在比较稳定的条件下制造。在与此相比不饱和脂肪族一元羧酸(b)与芳香族一元羧酸(c)的合计加入量多的情况下,具有羧基的不饱和脂肪族一元羧酸(b)、芳香族一元羧酸(c)残存,固化后的涂膜的耐溶剂性降低,因此不优选。另外,在过少的情况下,未反应的环氧化合物(a)残留,因此有时所得的聚氨酯的稳定性降低。更优选为0.95~1.15当量,进一步优选为1.0~1.1当量。此外,相对于不饱和脂肪族一元羧酸(b)与芳香族一元羧酸(c)的合计,芳香族一元羧酸(c)的比例优选为20~80摩尔%,更优选为25~75摩尔%。如果芳香族一元羧酸(c)为20摩尔%以上,则能够抑制最终获得的聚氨酯的固化收缩,能够减小基材的翘曲。此外,如果为80摩尔%以下,则通过基于(甲基)丙烯酰基的固化反应而表现良好的耐溶剂性。羧酸酯化反应可以在无溶剂的条件下反应或用溶剂进行稀释而反应。在使用溶剂的情况下,只要是对羧酸酯化反应不产生影响的溶剂,就没有特别限定。在使用溶剂的情况下,作为其使用量,应该根据生成物的粘度、用途而适当调整,但优选只要以固体成分含有率成为99~30质量%,更优选成为99~45质量%的方式使用即可。在反应所使用的化合物为高粘度的情况下粘度被抑制,适合反应进行。作为上述溶剂,特别优选为例如,酯系溶剂、醚系溶剂、酮系溶剂等能够在接下来的工序的制造氨基甲酸酯树脂时使用的溶剂。作为上述酯系溶剂,可举出例如,乙酸乙酯、乙酸丙酯、乙酸丁酯等乙酸烷基酯类、γ-丁内酯等环状酯类、乙二醇单甲基醚单乙酸酯、二甘醇单甲基醚单乙酸酯、二甘醇单乙基醚单乙酸酯、三甘醇单乙基醚单乙酸酯、二甘醇单丁基醚单乙酸酯、丙二醇单甲基醚单乙酸酯、丁二醇单甲基醚单乙酸酯等单亚烷基二醇单烷基醚单乙酸酯类或聚亚烷基二醇单烷基醚单乙酸酯类。作为上述醚系溶剂,可举出例如,乙醚、乙基丁基醚等烷基醚类、乙二醇二甲基醚、乙二醇二乙基醚、双丙甘醇二甲基醚、双丙甘醇二乙基醚、三甘醇二甲基醚、三甘醇二丁基醚等二醇醚类、四氢呋喃等环状醚类等。作为上述酮系溶剂,可举出例如,丙酮、甲基乙基酮、环己酮、异佛尔酮等。其中,更优选为沸点为80℃以上且350℃以下的溶剂,进一步优选为沸点为100℃以上且300℃以下的溶剂。如果沸点为80℃以上,则溶剂的沸点越高,反应温度越高,以使羧酸酯化反应顺利进行。此外,溶剂的温度不过高时(350℃以下),在以后的工序中,能够在更短时间除去溶剂,因此工业上是有利的。在羧酸酯化反应中,为了促进反应,优选使用催化剂。在使用该催化剂的情况下,其使用量相对于环氧化合物(a)、不饱和脂肪族一元羧酸(b)和芳香族一元羧酸(c)的总量、在使用溶剂的情况下还加入了溶剂的总量为0.1~3质量%左右。此时的反应温度为60~150℃,反应时间优选为5~60小时。作为该催化剂,优选为不含卤素的催化剂,可举出例如,三乙胺、苄基二甲基胺、三苯基膦、辛酸锆等一般的碱性催化剂等。此外,可以使用热阻聚剂,作为该热阻聚剂,优选使用例如,吩噻嗪、氢醌单甲基醚、2-甲基氢醌、氢醌、二苯基苦味酰肼(diphenylpicrylhydrazine)、二苯基胺、3,5-二-叔丁基-4-羟基甲苯等。羧酸酯化反应中,一边适当取样,一边将样品的酸值变为5mg·koh/g以下,优选变为2mg·koh/g以下的时刻设为终点。在本说明书中,酸值是通过以下方法测定的值。在100ml锥形烧瓶中利用精密天平精密称量试样约0.2g,在其中加入乙醇/甲苯=1/2(质量比)的混合溶剂10ml进行溶解。进一步,在该容器中添加作为指示剂的酚酞乙醇溶液1~3滴,充分搅拌直到试样变得均匀为止。将其用0.1n氢氧化钾-乙醇溶液滴定,将指示剂的微红色持续30秒的时刻作为中和的终点。将由该结果使用下述计算式计算获得的值设为树脂的酸值。酸值(mgkoh/g)=〔b×f×5.611〕/sb:0.1n氢氧化钾-乙醇溶液的使用量(ml)f:0.1n氢氧化钾-乙醇溶液的因子s:试样的采取量(g)在以上说明的环氧羧酸酯化合物(x)的合成中,如果使作为羧酸成分的作为不饱和脂肪族一元羧酸(b)的(甲基)丙烯酸与芳香族一元羧酸(c)的混合物、与作为环氧化合物(a)的芳香族二缩水甘油基醚进行反应,则生成3种化合物,生成至少包含式(1)所示结构的第一环氧羧酸酯化合物和第二环氧羧酸酯化合物的环氧羧酸酯组合物。式(1)中,a为来源于二缩水甘油基醚的有机残基,且不具有羟基。r1、r2各自独立地为可以具有取代基的碳原子数为6~30的芳香环或式(2)所示的有机基团,上述第一环氧羧酸酯化合物的r1、r2中的至少一个为可以具有取代基的碳原子数为6~30的芳香环,上述第二环氧羧酸酯化合物的r1、r2中的至少一个为式(2)所示的有机基团,第一环氧羧酸酯化合物与第二环氧羧酸酯化合物不同。上述可以具有取代基的碳原子数为6~30的芳香环与芳香族一元羧酸(c)所具有的碳原子数为6~30的芳香环对应。即,可举出可以具有不含烯属不饱和基的取代基、具体为碳原子数为1~6的烷基、碳原子数1~6的烷氧基等作为取代基的、苯环或萘骨架、蒽骨架等稠环等。h2c=cr3-(2)式(2)中,r3表示氢原子或甲基。更具体而言,作为环氧化合物(a),使用了双酚a-二缩水甘油基醚,作为不饱和脂肪族一元羧酸(b),使用了丙烯酸,作为芳香族一元羧酸(c),使用了苯甲酸的情况下的反应如下所述。所得的环氧羧酸酯化合物(a)包含2个缩水甘油基醚基都与丙烯酸反应了的化合物1、2个缩水甘油基醚基都与苯甲酸反应了的化合物2、和2个缩水甘油基醚基中的一个与丙烯酸反应且另一个与苯甲酸反应了的化合物3。通过使包含它们的环氧羧酸酯化合物(x)与后述的二异氰酸酯化合物(y)进行反应,从而获得包含化合物1(相当于第二环氧羧酸酯化合物)与二异氰酸酯化合物(y)的第二氨基甲酸酯键单元、化合物2(相当于第一环氧羧酸酯化合物)与二异氰酸酯化合物(y)的第一氨基甲酸酯键单元、和化合物3(相当于第一或第二环氧羧酸酯化合物)与二异氰酸酯化合物(y)的第三氨基甲酸酯键单元的聚氨酯。通过将作为二羟基化合物的羧酸酯骨架包含芳香环的环氧羧酸酯化合物(x)用于后述的聚氨酯的合成而最终获得的聚氨酯、与聚对苯二甲酸乙二醇酯(pet)、聚萘二甲酸乙二醇酯(pen)、芳香族聚酰亚胺(pi)等分子中具有芳香环的基材的密合性提高,并且聚氨酯的耐固化收缩性也可以提高。<聚氨酯的合成:第二工序>作为上述二异氰酸酯化合物(y),可举出例如,脂肪族二异氰酸酯、脂环族二异氰酸酯、芳香族二异氰酸酯、芳香脂肪族二异氰酸酯等。在合成的聚氨酯不凝胶化的范围内,也可以少量使用三苯基甲烷三异氰酸酯那样的、具有3个以上异氰酸酯基的多异氰酸酯。作为脂肪族二异氰酸酯,可举出例如,1,3-丙二异氰酸酯、1,4-丁二异氰酸酯、1,6-己二异氰酸酯、1,9-壬二异氰酸酯、1,10-癸二异氰酸酯、2,2,4-三甲基六亚甲基二异氰酸酯、2,4,4-三甲基六亚甲基二异氰酸酯、赖氨酸二异氰酸酯、2,2’-二乙基醚二异氰酸酯、二聚酸二异氰酸酯等。作为脂环族二异氰酸酯,可举出例如,1,4-环己烷二异氰酸酯、1,3-双(异氰酸酯基甲基)环己烷、1,4-双(异氰酸酯基甲基)环己烷、3-异氰酸酯基甲基-3,5,5-三甲基环己基异氰酸酯(ipdi,异佛尔酮二异氰酸酯)、双-(4-异氰酸酯基环己基)甲烷(二环己基甲烷4,4’-二异氰酸酯,氢化mdi)、氢化(1,3-或1,4-)苯二亚甲基二异氰酸酯、降冰片烷二异氰酸酯、亚甲基双(4-环己基异氰酸酯)等。作为芳香族二异氰酸酯,可举出例如,2,4’-二苯基甲烷二异氰酸酯、4,4’-二苯基甲烷二异氰酸酯、1,4-苯二异氰酸酯、2,4-甲苯二异氰酸酯、2,6-甲苯二异氰酸酯、(1,2、1,3或1,4)-二甲苯二异氰酸酯、3,3’-二甲基-4,4’-二异氰酸酯基联苯、3,3’-二甲基-4,4’-二异氰酸酯基二苯基甲烷、1,5-萘二异氰酸酯、4,4’-二苯基醚二异氰酸酯、四氯苯二异氰酸酯等。作为芳香脂肪族二异氰酸酯,可举出例如,1,3-苯二亚甲基二异氰酸酯,1,4-苯二亚甲基二异氰酸酯、α,α,α’,α’-四甲基苯二亚甲基二异氰酸酯、3,3’-亚甲基二甲代亚苯基-4,4’-二异氰酸酯(3,3'-methyleneditolylene-4,4'-diisocyanate)等。这些二异氰酸酯可以单独使用1种或组合使用2种以上。通过使用除异氰酸酯基(-nco基)中的碳原子以外的碳原子的数为6~30的脂环式化合物作为二异氰酸酯化合物(y),从而由后述的实施例涉及的聚氨酯树脂形成的保护膜特别是在高温高湿时的可靠性高,适合电子设备部件的构件。期望上述脂环式化合物,在二异氰酸酯化合物(y)中,相对于二异氰酸酯化合物(y)的总量(100mol%)包含30mol%以上,优选包含50mol%以上,进一步优选包含70mol%以上。本实施方式的聚氨酯可以仅由上述成分(x)和(y)合成,但出于对该聚氨酯进一步赋予自由基聚合性、阳离子聚合性的目的,或抑制聚氨酯末端的异氰酸酯基、羟基的残基的影响的目的,可以进一步使(z)单羟基化合物和/或(w)单异氰酸酯化合物反应而合成。单羟基化合物(z)作为单羟基化合物(z),可举出(甲基)丙烯酸2-羟基乙酯、(甲基)丙烯酸羟基丙酯、(甲基)丙烯酸羟基丁酯、环己烷二甲醇单(甲基)丙烯酸酯、上述各(甲基)丙烯酸酯的己内酯或氧化烯加成物、甘油二(甲基)丙烯酸酯、三羟甲基二(甲基)丙烯酸酯、季戊四醇三(甲基)丙烯酸酯、二季戊四醇五(甲基)丙烯酸酯、双三羟甲基丙烷三(甲基)丙烯酸酯、烯丙醇、烯丙基氧基乙醇等具有自由基聚合性双键的化合物、乙醇酸、羟基新戊酸等具有羧酸的化合物。单羟基化合物(z)可以单独使用1种或组合使用2种以上。此外,这些化合物中,优选为(甲基)丙烯酸2-羟基乙酯、(甲基)丙烯酸羟基丙酯、(甲基)丙烯酸羟基丁酯、烯丙醇、乙醇酸、羟基新戊酸,更优选为(甲基)丙烯酸2-羟基乙酯和(甲基)丙烯酸4-羟基丁酯。此外,作为单羟基化合物(z),可举出甲醇、乙醇、正丙醇、异丙醇、正丁醇、异丁醇、仲丁醇、叔丁醇、戊醇、己醇、辛醇等。单异氰酸酯化合物(w)作为单异氰酸酯化合物(w),可举出(甲基)丙烯酰氧基乙基异氰酸酯、异氰酸苯酯、异氰酸己酯、异氰酸十二烷基酯、カレンズmoi(注册商标)、カレンズaoi(注册商标)、カレンズbei(注册商标)(昭和电工制)等。实施方式涉及的聚氨酯可以如下合成:通过在存在或不存在二月桂酸二丁基锡、四乙酰丙酮锆等金属催化剂那样的公知的氨基甲酸酯化催化剂的条件下,使用适当的有机溶剂,使上述环氧羧酸酯化合物(x)与二异氰酸酯化合物(y),根据需要并用单羟基化合物(z)和/或单异氰酸酯化合物(w)进行反应来合成。上述有机溶剂只要是与异氰酸酯化合物反应性低的有机溶剂,就没有特别限定,优选为胺等不含碱性官能团的溶剂。作为这样的溶剂,可以举出例如,甲苯、二甲苯、乙基苯、硝基苯、环己烷、异佛尔酮、二甘醇二甲基醚、乙二醇二乙基醚、乙二醇单甲基醚单乙酸酯、丙二醇单甲基醚单乙酸酯、丙二醇单乙基醚单乙酸酯、双丙甘醇单甲基醚单乙酸酯、二甘醇单乙基醚单乙酸酯、甲氧基丙酸甲酯、甲氧基丙酸乙酯、乙氧基丙酸甲酯、乙氧基丙酸乙酯、乙酸乙酯、乙酸正丁酯、乙酸异戊酯、乳酸乙酯、丙酮、甲基乙基酮、环己酮、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n-甲基吡咯烷酮、γ-丁内酯、二甲亚砜、氯仿和二氯甲烷等。另外,由于不优选生成的聚氨酯的溶解性低的有机溶剂,因此其中,特别优选为丙二醇单甲基醚单乙酸酯、丙二醇单乙基醚单乙酸酯、双丙甘醇单甲基醚单乙酸酯、二甘醇单乙基醚单乙酸酯、γ-丁内酯等。对进行原料加入的顺序没有特别限制,通常将环氧羧酸酯化合物(x)先加入到反应容器中,使其溶解于溶剂后,在20~150℃、更优选为60~120℃下,一边滴加二异氰酸酯化合物(y)一边加入,然后,在30~160℃、更优选为50~130℃下通常反应2~15小时。另外,反应可以在大气气氛下实施,也可以在氮气等非活性气氛下进行。原料的加入摩尔比根据作为目标的聚氨酯的分子量和酸值来调节。具体而言,它们的加入摩尔比是,(二异氰酸酯化合物(y)的异氰酸酯基):(环氧羧酸酯化合物(x)的羟基)为0.5~1.5:1,优选为0.8~1.2:1,更优选为0.95~1.05:1。在向聚氨酯导入单羟基化合物(z)的情况下,为了使聚氨酯分子的末端成为异氰酸酯基,需要使用与环氧羧酸酯化合物(x)相比过剩的二异氰酸酯化合物(y)(与羟基的合计相比异氰酸酯基变为过剩的方式)。在使用单羟基化合物(z)的情况下,优选使二异氰酸酯化合物(y)的摩尔数与环氧羧酸酯化合物(x)的摩尔数相比过剩,以相对于异氰酸酯基的过剩摩尔数为0.5~1.5倍摩尔量、优选为0.8~1.2倍摩尔量使用单羟基化合物(z)。如果单羟基化合物(z)的摩尔量为0.5倍摩尔量以上,则能够降低聚氨酯的末端的异氰酸酯基,如果为1.5倍摩尔量以下,则能够防止未反应的单羟基化合物(z)残存而对后面工序产生不良影响。为了将单羟基化合物(z)导入到聚氨酯,在环氧羧酸酯化合物(x)与二异氰酸酯化合物(y)的反应基本上结束的时刻,为了使聚氨酯的两末端残存的异氰酸酯基与单羟基化合物(z)反应,在反应溶液中在20~150℃,更优选为在70~120℃下滴加单羟基化合物(z),然后在该温度下保持而使反应结束。如果滴加和反应温度为20℃以上,则残存的异氰酸酯基与单羟基的反应迅速进行,在150℃以下的情况下,能够防止滴加时反应迅速进行而失控。在使用单异氰酸酯化合物(w)的情况下,优选使环氧羧酸酯化合物(x)的摩尔数与二异氰酸酯化合物(y)的摩尔数相比过剩,相对于羟基的过剩摩尔数以0.5~1.5倍摩尔量,优选以0.8~1.2倍摩尔量使用。如果单异氰酸酯化合物(w)的摩尔量为0.5倍摩尔量以上,则能够减少聚氨酯的末端的羟基,如果为1.5倍摩尔量以下,则能够防止未反应的单异氰酸酯化合物(w)残存而对后面工序产生不良影响。为了将单异氰酸酯化合物(w)导入到聚氨酯,在环氧羧酸酯化合物(x)与二异氰酸酯化合物(y)的反应基本上结束的时刻,为了使聚氨酯的两末端残存的羟基与单异氰酸酯化合物(w)反应,在反应溶液中在20~150℃、更优选为在50~120℃下滴加单异氰酸酯化合物(w),然后在该温度下保持而使反应结束。如果滴加和反应温度为20℃以上,则残存的羟基与单异氰酸酯化合物(w)的反应迅速进行,在150℃以下的情况下,能够防止滴加时反应迅速进行而失控。如以上那样操作而合成的聚氨酯的重均分子量为5000~40000是适合的。重均分子量是通过以下说明的方法通过gpc测定的值。在本说明书中,只要没有特别指明,gpc的测定条件如下所述。装置名:日本分光株式会社制hplc单元hss-2000柱:shodex柱lf-804流动相:四氢呋喃流速:1.0ml/min检测器:日本分光株式会社制ri-2031plus温度:40.0℃试样量:进样环管100μl试样浓度:调制成约0.1质量%<聚氨酯树脂组合物>通过在上述聚氨酯中添加光引发剂、和根据需要的其它具有自由基聚合性基团的单体类、更优选为单官能和多官能丙烯酸酯,从而获得uv固化性的聚氨酯树脂组合物。该单体类的添加量相对于聚氨酯100质量份为50~300质量份,优选为80~200质量份。配合于uv固化性的聚氨酯树脂组合物的光引发剂的配合量不特别规定,但优选相对于聚氨酯(在包含其它具有自由基聚合性基团的单体类的情况下为聚氨酯与其它具有自由基聚合性基团的单体类的总和)100质量份为0.5~15质量份,优选为1~10质量份。如果多于15质量份,则在uv固化后大量的光引发剂残留,成为污染的原因。此外,如果小于0.5质量份,则可能会在uv照射时未进行充分的反应而固化不足,粘着力不降低,发生拾取(pickup)不良状况等。作为光引发剂,没有特别限定,但从对紫外线的反应性高方面考虑,优选使用光自由基引发剂。作为光自由基引发剂,可举出例如,苯乙酮、苯丙酮、二苯甲酮、吨醇(キサントール)、芴、苯甲醛、蒽醌、三苯基胺、咔唑、3-甲基苯乙酮、4-甲基苯乙酮、3-戊基苯乙酮、2,2-二乙氧基苯乙酮、4-甲氧基苯乙酮、3-溴苯乙酮、4-烯丙基苯乙酮、对二乙酰苯、3-甲氧基二苯甲酮、4-甲基二苯甲酮、4-氯二苯甲酮、4,4’-二甲氧基二苯甲酮、4-氯-4’-苄基二苯甲酮、3-氯吨酮、3,9-二氯吨酮、3-氯-8-壬基吨酮、苯偶姻、苯偶姻甲基醚、苯偶姻丁基醚、双(4-二甲基氨基苯基)酮、苄基甲氧基缩酮、2-氯噻吨酮、2,2-二甲氧基-1,2-二苯基乙烷-1-酮(irgacure(注册商标)651,basfジャパン制)、1-羟基-环己基-苯基-酮(irgacure(注册商标)184,basfジャパン制)、2-羟基-2-甲基-1-苯基-丙烷-1-酮(darocur(注册商标)1173,basfジャパン制)、1-[4-(2-羟基乙氧基)-苯基]-2-羟基-2-甲基-1-丙烷-1-酮(irgacure(注册商标)2959,basfジャパン制)、2-甲基-1-[4-(甲硫基)苯基]-2-吗啉代丙烷-1-酮(irgacure(注册商标)907,basfジャパン制)、2-苄基-2-二甲基氨基-1-(4-吗啉代苯基)-丁酮-1(irgacure(注册商标)369,basfジャパン制)、2-(4-甲基苄基)-2-二甲基氨基-1-(4-吗啉-4-基-苯基)-丁烷-1-酮(irgacure(注册商标)379,basfジャパン制)、二苯甲酰(dibenzoyl)等。其中,优选为α-羟基酮化合物(例如,苯偶姻、苯偶姻甲基醚、苯偶姻丁基醚、1-羟基-环己基-苯基-酮等)、苯基酮衍生物(例如,苯乙酮、苯丙酮、二苯甲酮、3-甲基苯乙酮、4-甲基苯乙酮、3-戊基苯乙酮、2,2-二乙氧基苯乙酮、4-甲氧基苯乙酮、3-溴苯乙酮、4-烯丙基苯乙酮、3-甲氧基二苯甲酮、4-甲基二苯甲酮、4-氯二苯甲酮、4,4’-二甲氧基二苯甲酮、4-氯-4’-苄基二苯甲酮、双(4-二甲基氨基苯基)酮等)。进一步,作为能够抑制固化物表面的氧抑制的引发剂种类,可以使用分子内具有2个以上光分解性基团的光自由基引发剂、分子内具有3个以上芳香环的夺氢型光自由基引发剂。作为分子内具有2个以上光分解性基团的光自由基引发剂,可举出2-羟基-1-[4-[4-(2-羟基-2-甲基-丙酰基)-苄基]苯基]-2-甲基-丙烷-1-酮(irgacure(注册商标)127,basfジャパン制)、1-〔4-(4-苯甲酰苯基硫烷基)苯基〕-2-甲基-2-(4-甲基苯基磺酰基)丙烷-1-酮(商品名:esacure1001mlamberti制)、苯甲酰甲酸甲酯(speedcure(注册商标)mbflambson制)o-乙氧基亚氨基-1-苯基丙烷-1-酮(speedcure(注册商标)pdolambson制)、寡聚[2-羟基-2-甲基-[4-(1-甲基乙烯基)苯基]丙酮(商品名:esacurekip150lamberti制)等。作为分子内具有3个以上芳香环的夺氢型光自由基引发剂,可举出〔1-(4-苯基硫烷基苯甲酰基)亚庚基氨基〕苯甲酸酯、〔1-[9-乙基-6-(2-甲基苯甲酰基)咔唑-3-基]亚乙基氨基〕乙酸酯、4-苯甲酰基-4’甲基二苯硫醚、4-苯基二苯甲酮、4,4’,4”-(六甲基三氨基)三苯基甲烷等。此外,可以使用以改善深部固化性作为特征的光自由基引发剂。作为以改善深部固化性作为特征的光自由基引发剂,可举出2,4,6-三甲基苯甲酰基-二苯基-氧化膦(darocur(注册商标)tpo、basfジャパン制)、双(2,4,6-三甲基苯甲酰基)-苯基氧化膦(irgacure(注册商标)819、basfジャパン制)、双(2,6-二甲基苯甲酰基)-2,4,4-三甲基-戊基氧化膦等酰基氧化膦系光自由基引发剂。作为光自由基引发剂,在本发明的聚氨酯树脂组合物的固化性与储存稳定性的平衡方面,更优选为1-羟基-环己基-苯基-酮(irgacure(注册商标)184,basfジャパン制)、2-羟基-2-甲基-1-苯基-丙烷-1-酮(darocur(注册商标)1173,basfジャパン制)、双(4-二甲基氨基苯基)酮、2-羟基-1-[4-[4-(2-羟基-2-甲基-丙酰基)-苄基]苯基]-2-甲基-丙烷-1-酮(irgacure(注册商标)127,basfジャパン制)、2-苄基-2-二甲基氨基-1-(4-吗啉代苯基)-丁酮-1(irgacure(注册商标)369,basfジャパン制)、2-(4-甲基苄基)-2-二甲基氨基-1-(4-吗啉-4-基-苯基)-丁烷-1-酮(irgacure(注册商标)379,basfジャパン制)、2,4,6-三甲基苯甲酰基-二苯基-氧化膦(darocur(注册商标)tpo,basfジャパン制)、双(2,4,6-三甲基苯甲酰基)-苯基氧化膦(irgacure(注册商标)819,basfジャパン制)、双(2,6-二甲基苯甲酰基)-2,4,4-三甲基-戊基氧化膦。这些光自由基引发剂可以单独使用或混合使用2种以上,也可以与其它化合物组合使用。作为与其它化合物的组合,具体而言,可举出与4,4’-双(二甲基氨基)二苯甲酮、4,4’-双(二乙基氨基)二苯甲酮、二乙醇甲基胺、二甲基乙醇胺、三乙醇胺、乙基-4-二甲基氨基苯甲酸酯、2-乙基己基-4-二甲基氨基苯甲酸酯等胺的组合、进一步在其中组合了二苯基碘氯化物等碘盐的组合、与亚甲基蓝等色素和胺的组合等。另外,在使用上述光自由基引发剂的情况下,根据需要,也可以添加氢醌、氢醌单甲基醚、苯醌、对叔丁基儿茶酚等阻聚剂类。此外,在uv固化性的聚氨酯树脂组合物中也可以配合光敏化剂。作为光敏化剂,可举出三乙胺、三正丁基膦等。进而,也可以在uv固化性的聚氨酯树脂组合物中混合热固化引发剂。作为热固化引发剂,可以使用偶氮系、过氧化物系等以往公知的热固化引发剂。另外,在通过uv进行树脂层的聚合的情况下,一般而言适量添加光引发剂,此外也可以根据需要适量添加光敏化剂。作为该光引发剂,可举出苯乙酮、二苯甲酮、苯偶姻、苯甲酸苯甲酰酯、噻吨酮类等。作为光敏化剂,可举出三乙胺、三正丁基膦等。实施例以下具体地说明本发明的实施例。另外,以下实施例是为了使本发明的理解容易,本发明不限制于这些实施例。合成例1在500ml三口圆底烧瓶中,加入将碳酸钾(日本曹达株式会社制)138g溶解于纯水125g的溶液、双酚a(东京化成工业株式会社制)228.29g、和碳酸钠(关东化学株式会社制)52.0g(0.500mol,固体状态),将反应器进行氮气置换并加热到85℃。在氮气气流下,加入乙酸烯丙酯(昭和电工株式会社制)220g、三苯基膦(北兴化学株式会社制)2.62g、和50%含水5%-pd/c-std型(エヌ·イーケムキャット株式会社制)84.6mg,在氮气气氛中,升温到105℃反应4小时,然后追加乙酸烯丙酯22.0g,继续加热12小时。在反应结束后,将反应体系冷却直到室温,然后加入纯水直到析出的盐全部溶解,进行了分液处理。分离出有机层,将有机溶剂(70℃,50mmhg,2小时)蒸馏除去。添加纯水(200g)后,加入甲苯200g,保持在80℃以上的温度而确认了没有析出白色沉淀,然后通过过滤(1微米的膜滤器(使用アドバンテック社制kst-142-ja进行加压(0.3mpa))来回收pd/c。将该滤渣用甲苯100g进行洗涤,并且分离出水层。在50℃以上将有机层用纯水200g洗涤2次,确认了水层为中性。分离出有机层后,在减压下进行浓缩,获得了以双酚a二烯丙基醚作为主成分的淡黄色液体(283.76g,92.0%收率)。将所得的双酚a二烯丙基醚95.7g、甲醇259.0g、乙腈130.9g加入到烧瓶中,进行了搅拌。将过氧化氢(35质量%)399.9g从滴液漏斗以约40g/hr的速度滴加。加热直到40℃,添加碳酸钾水溶液(50质量%),一边以ph值成为10~11的方式调整一边在30~40℃反应12小时。通过蒸发器将甲醇和残留乙腈等蒸馏除去。在分液漏斗中加入反应液、甲苯1.2升、10%硫酸钠水溶液120ml并进行了分液处理。用纯水200ml进行洗涤分液处理后,在有机层中加入2%亚硫酸钠水溶液200ml进行了分液处理。水层的ph值为9,用碘化钾淀粉纸确认了为无色,然后在有机层中加入纯水200ml再次进行了分液处理,确认了水层为ph=7后,将有机层进行减压浓缩直到甲苯浓度变为1%以下。另外,甲苯浓度通过气相色谱确认,合成出无卤素的双酚-a二缩水甘油基醚(以下,表述为hf-badg)。图1中表示所得的环氧值162的hf-badg的1h-nmr光谱。合成例2(hf-badg丙烯酸酯的合成)在室温下将合成例1中合成的hf-badg100g(0.29摩尔)、吩噻嗪(和光纯药工业(株)制)0.4g、三苯基膦(北兴化学工业(株)制)0.4g加入烧瓶中,一边搅拌,然后一边以约5℃/分钟的速度升温到70℃。在其中慢慢地滴加丙烯酸(昭和电工(株)制)41.3g(0.57摩尔)。在滴加结束后以约5℃/分钟的速度升温到120℃,保持120℃。在反应中继续搅拌。适时取样,测定了酸值。将酸值变为5以下的时刻设为反应终点。另外,一系列反应在大气气氛下进行。收量为126.6g,收率为89.8%,酸值为4.4,粘度为504pa·s。通过1h-nmr进行所得的化合物的鉴定。将结果示于图2中。与图1相比,2.8ppm附近的来源于环氧基的峰消失,在6.0~6.5ppm附近可以新确认到来源于丙烯酰基的峰,因此确认了badg丙烯酸酯的合成。合成例3~7:第一工序合成例3~5、7中,如表1所记载那样代替丙烯酸而使用丙烯酸与苯甲酸的混合物(合成例3~5)、或苯甲酸(合成例7),将反应规模变更为1/2进行合成,除此以外,与合成例2同样地实施。合成例6中,代替合成例1中合成的hf-badg而使用市售品(东京化成工业(株)制2,2-双(4-缩水甘油基氧基苯基)丙烷(双酚-a二缩水甘油基醚(badg))),将反应规模变更为1/2进行合成,除此以外,与合成例2同样地实施。以下,以合成例3为例进行说明。将合成例1中合成的hf-badg50.0g、苯甲酸(关东化学(株)制)17.45g、吩噻嗪(和光纯药工业(株)制)0.2g、三苯基膦(北兴化学工业(株)制)0.3g、丙二醇单甲基醚乙酸酯10.4g加入到烧瓶中,在70℃下搅拌。在其中慢慢地滴加丙烯酸(昭和电工(株)制)10.3g。滴加后升温到120℃,进行了搅拌。适时取样,测定了酸值。将酸值变为5以下的时刻设为反应终点。收量为77.2g,收率为88%,酸值为4.0,固体浓度为91.2质量%,粘度为510pa·s。与合成例3同样地合成的合成例4的收量为74.0g,收率为87%,酸值为2.0,固体浓度为90.2质量%,粘度为550pa·s,合成例5的收量为82.1g,收率为89%,酸值为1.9,固体浓度为90.6质量%,粘度为520pa·s,合成例6的收量为73.4g,收率为90%,酸值为1.7,固体浓度为89.8质量%,粘度为1200pa·s,合成例7的收量为84.4g,收率为88%,酸值为3.8,固体浓度为91.1质量%,粘度为570pa·s。[表1]badg丙烯酸(g)苯甲酸(g)合成例2合成例141.3合成例3合成例110.317.45合成例4合成例115.458.73合成例5合成例15.1526.19合成例6市售品20.65合成例7合成例135市售品:东京化成工业(株)制2,2-双(4-缩水甘油基氧基苯基)丙烷另外,关于合成例6中制造的badg丙烯酸酯,也与合成例2同样地,通过1h-nmr进行了化合物的鉴定。将结果示于图3中。与图2(合成例2的badg丙烯酸酯)的峰相比,2.7~3.4ppm附近的峰不同。此外,与由合成例1的badg进行了羧酸酯化的各合成例相比,粘度显著变高。由此,暗示出合成例6中获得的badg丙烯酸酯中具有伴随环氧基的羧酸酯化而生成的羟基以外的羟基。可以认为这是因为,合成例6中使用的badg(市售品)中包含后述的杂质(式(4))。实施例1(聚氨酯的合成;第二工序)将合成例3(第一工序)中合成的hf-badg丙烯酸酯12.3g、作为反应催化剂的オルガチツクスzc150(四乙酰丙酮锆,マシモトファィングミ力ル社制)0.2g、丙二醇单甲基醚乙酸酯15.9g在室温下加入到具备搅拌装置、温度计、冷凝器的300ml三口烧瓶中后进行搅拌,一边流通氮气一边以约5℃/分钟升温到90℃,进行保持。在其中慢慢地滴加二环己基甲烷4,4’-二异氰酸酯(デスモジコ一ル(注册商标)w(住化コベストロウレタン(株)制)5.5g,在滴加结束后以约5℃/分钟升温到110℃后在110℃保持,继续反应5小时。在反应中进行搅拌,继续氮气流动。所得的聚氨酯1的固体浓度为55.3%,重均分子量为14000。其它实施例2、3、比较例1、2、4中,将实施例1中使用的合成例3的hf-badg丙烯酸酯和其质量如表2所记载那样变更,除此以外,与实施例1同样地实施。另外,比较例3中使用的badg(双酚-a二缩水甘油基醚)丙烯酸酯为大阪有机化学工业(株)制ビスコート#540。实施例2中获得的聚氨酯2的固体浓度为54.2%,重均分子量为13500,实施例3中获得的聚氨酯3的固体浓度为55.5%,重均分子量为15000,比较例2中获得的聚氨酯4的固体浓度为53.8%,重均分子量为16000,比较例4中获得的聚氨酯5的固体浓度为52.1%,重均分子量为14500。[表2]实施例1~3、比较例2、4中,未凝胶化,获得了聚氨酯。比较例1、3中,凝胶化,不能获得聚氨酯。一般而言,市售品的badg以表氯醇和双酚a作为原料而合成,市售品的badg中包含作为此时的副产物的下述式(4)所示的二聚体。因此,比较例1中使用的badg丙烯酸酯中,包含含有3个羟基的下述式(5)所示的杂质。其结果可以认为,第二工序中的加聚反应时发生交联反应,凝胶化而得不到链状的聚氨酯。上述式(5)的杂质由图3所示的1h-nmr光谱确认。此外明示出,比较例3中使用的市售的badg丙烯酸酯包含二聚体,可以认为通过含有羟基为3个的化合物,同样地伴随凝胶化。此外,比较例4中,由于使用与比较例1、比较例3中使用的丙烯酸酯主成分相同,但在合成例2中获得的hf-badg,因此原料badg中包含的副产物少,在氨基甲酸酯化时不凝胶化。对以上的实施例1~3、比较例2、4中获得的聚氨酯进行了涂膜评价。将结果示于表3中。将各聚氨酯利用丙二醇单甲基醚乙酸酯,以用e型粘度计测定的25℃下的粘度成为3000mpa·s左右的方式进行稀释,相对于所得的稀释品100质量份添加光引发剂irgacure(注册商标)184(basf社制)5质量份。使用自转/公转真空混合机あわとり練太郎(注册商标)arv-310(株式会社シンキー制)充分混合(以自转500圈、公转1500圈进行5分钟),制成涂布用的墨。作为基材使用pet(ルミラー(注册商标)t60(東レ社制)),利用棒式涂布机(no20)在该膜上涂布各墨,在80℃下干燥30分钟,然后在氮气气氛下用高压水银灯(波长365nm下的照度18mw/cm2)曝光120秒,使聚氨酯固化,在膜上形成作为固化物的固化膜。固化膜的膜厚为约15μm。氨基甲酸酯化:○:可以没有问题地合成×:凝胶化固化后密合性:依照jisk7600,利用划方格法实施并通过以下基准来判定。○:剥离试验中没有剥离△:一部分剥离×:全部剥离固化后翘曲:将被覆了固化膜的膜切出成100mm的正方形,使印刷面在上放置,通过以下基准进行了评价。○:端部与水平部的差2mm以内△:5mm以内×:超过5mm固化后丙酮耐性:在固化膜上,将丙酮用吸管滴1ml,5分钟后用抹布擦去,风干后,通过以下基准判定涂膜的外观。○:无溶解△:有一部分变质(白浊等)×:溶解[表3]另外,在表3中,比较例1和3中的固化后密合性、固化后翘曲、固化后丙酮耐性的栏的“-”是指,由于不能获得聚氨酯因此未评价。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1