从头组装条码化的基因组DNA片段的方法与流程
本申请要求于2016年8月10日提交的美国临时申请号62/373,057号的优先权,其通过引用纳入本文用于所有目的。
政府权益的声明
本发明是在国立卫生研究院(nationalinstitutesofhealth)的5dp1ca186693下于政府资助下完成。政府对本发明享有某些权利。
背景技术:
发明领域
本发明的实施方式通常涉及用于由单个细胞从头组装基因组核酸(如dna)的方法和组合物。
背景技术
基因组从头组装是在没有参照序列的帮助下将单个短测序读数组装成较长序列的过程。当前,大多数的高通量测序产生仅几百个碱基对的序列长度。然后,通过确定这些片段在何处重叠,将这些短片段重建在一起。然而,在复杂的生物体,例如人类的基因组中存在许多重复序列。许多这些重复区域长于dna测序仪的读数长度,这使得组装没有缺口的全基因组十分困难。
在细胞间变异和种群异质性起关键作用的研究中,如肿瘤生长、干细胞重编程、胚胎发育等,进行单个细胞基因组测序的能力非常重要。当进行测序的细胞样品非常珍贵或稀有或以微量存在时,单个细胞基因组测序也同样非常重要。准确的单个细胞基因组测序的重要之处在于基因组dna(可以是处于微量的)的初始扩增。
扩增和测序后的基因组从头组装是用于全基因组测序的许多方法的一个重要方面。全基因组扩增方法包括多重置换扩增(mda),其是在测序和其它分析之前在采用来自单个细胞的基因组dna的领域的常用方法。在该方法中,随机引物退火后,利用具有强链置换活性的dna聚合酶进行延伸。来自单个细胞的原始基因组dna以级联样的形式指数地扩增,以形成超支化dna结构。扩增来自单个细胞的基因组dna的其它方法述于zong,c.,lu,s.,chapman,a.r.,和xie,x.s.(2012),单个人细胞的单核苷酸和拷贝数变异的基因组范围检测(genome-widedetectionofsingle-nucleotideandcopy-numbervariationsofasinglehumancell),science338,1622-1626,其中描述了多重退火和基于环型的扩增循环(malbac)。本领域所知的另一方法是简并寡核苷酸引物pcr或dop-pcr。用于单个细胞基因组dna的若干其他方法包括:cheung,v.g.和s.f.nelson,使用简并寡核苷酸的全基因组扩增允许成百上千个基因型以少于一纳克的基因组dna进行(wholegenomeamplificationusingadegenerateoligonucleotideprimerallowshundredsofgenotypestobeperformedonlessthanonenanogramofgenomicdna),proceedingsofthenationalacademyofsciencesoftheunitedstatesofamerica,1996.93(25):14676-9页;telenius,h.,等,简并寡核苷酸引发的pcr:通过单个简并引物的常规扩增(degenerateoligonucleotide-primedpcr:generalamplificationoftargetdnabyasingledegenerateprimer),genomics,1992.13(3):718-25页;zhang,l.,等,单个细胞的全基因组扩增:对基因分析的启示(wholegenomeamplificationfromasinglecell:implicationsforgeneticanalysis).proceedingsofthenationalacademyofsciencesoftheunitedstatesofamerica,1992,89(13):5847-51页;lao,k.,n.l.xu,和n.a.straus,使用单个引物的pcr的全基因组扩增(wholegenomeamplificationusingsingle-primerpcr),biotechnologyjournal,2008,3(3):378-82页;dean,f.b.,等,使用多重置换扩增的完整人基因组扩增(comprehensivehumangenomeamplificationusingmultipledisplacementamplification),proceedingsofthenationalacademyofsciencesoftheunitedstatesofamerica,2002.99(8):5261-6页;lage,j.m.,等,使用超支化链置换扩增和列阵-cgh对小dna样品中基因变异的全基因组分析(wholegenomeanalysisofgeneticalterationsinsmalldnasamplesusinghyperbranchedstranddisplacementamplificationandarray-cgh),genomeresearch,2003,13(2):294-307页;spits,c.,等,单个细胞全基因组置换扩增的优化和评价(optimizationandevaluationofsingle-cellwhole-genomemultipledisplacementamplification),humanmutation,2006,27(5):496-503页;gole,j.,等,使用纳米微微管进行单个细胞大规模平行聚合酶克隆和基因组测序(massivelyparallelpolymerasecloningandgenomesequencingofsinglecellsusingnanolitermicrowells),naturebiotechnology,2013.31(12):1126-32页;jiang,z.,等,使用多重置换扩增的单个精子的基因组扩增(genomeamplificationofsinglespermusingmultipledisplacementamplification),nucleicacidsresearch,2005,33(10):e91页;wang,j.,等,静止细胞中重组活性和新生突变比例的基因组范围单个细胞分析(genome-widesingle-cellanalysisofrecombinationactivityanddenovomutationratesinhumansperm),cell,2012.150(2):402-12页;ni,x.,肺癌患者单循环肿瘤细胞中可再生拷贝数变异模式(reproduciblecopynumbervariationpatternsamongsinglecirculatingtumorcellsoflungcancerpatients),pnas,2013,110,21082-21088;navin,n.,通过细胞测序推测肿瘤进化(tumorevolutioninferredbysinglecellsequencing),nature,2011,472(7341):90-94;evrony,g.d.,等,人大脑中11个反转录转座和体细胞突变的单个神经元测序分析(single-neuronsequencinganalysisofl1retrotranspositionandsomaticmutationinthehumanbrain),cell,2012.151(3):483-96页;和mclean,j.s.,等,使用高通量单个细胞基因组学平台从医院槽中的生物膜中回收牙龈卟啉单胞菌病原体的基因组(genomeofthepathogenporphyromonasgingivalisrecoveredfromabiofilminahospitalsinkusingahigh-throughputsingle-cellgenomicsplatform),genomeresearch,2013.23(5):867-77页。涉及全基因组扩增方面的方法报道于wo2012/166425,us7,718,403,us2003/0108870和us7,402,386。
然而,存在对于扩增少量基因组dna的其它方法的需求,诸如由其中扩增子可以被从头组装到基因组dna中的单个细胞或一小群细胞扩增。
技术实现要素:
本公开提供了用于基因组dna片段化的方法,其中在片段化过程期间,片段的邻接末端用相同的独特末端条码序列条码化,从而使经测序的片段后续可以通过连接具有相同的独特末端条码序列的片段被计算组装成较长的序列。根据一个方面,在水性介质中使用转座体(transposome)文库制造基因组dna的片段,其中将独特条码序列在被转座体的转座酶切割的位点处插入或连接基因组dna的各末端。本公开考虑了使用本文所述的转座体文库将基因组dna片段化成多个片段,如5或更多片段,10或更多片段,100或更多片段,1000或更多片段,10,000或更多片段,100,000或更多片段,1,000,000或更多片段,或10,000,000或更多片段。根据一个方面,转座体文库包含5-10个转座体成员,10-100个转座体成员,100或跟多个转座体成员,1000或更多个转座体成员,10,000或更多个转座体成员,100,000或更多个转座体成员,1,000,000或更多个转座体成员或10,000,00或更多个转座体成员。根据一个方面,各转座体包含2个转座酶和2个转座子dna。转座子dna包含转座酶结合位点,条码和引物结合位点。根据一个方面,转座子dna包含单个转座酶结合位点,条码和引物结合位点。各转座子dna是在转座酶结合位点与转座酶结合的单独的核酸。转座体是各自与其自身的转座子dna结合的两个单独的转座酶的二聚体。根据一个方面,转座体包含2个单独的和单个的转座子dna,各自结合其自身相应的转座酶。根据一个方面,转座体仅包含2个转座酶和2个转座子dna。根据一个方面,作为转座体部分的2个转座子dna是单独的、单个的或未连接的转座子dna,各自结合其自身相应的转座酶。例如,本文所述具有单个转座子结合位点,条码和引物结合位点的单独的和单个的转座子dna能够用于使用微滴方法制造数百万的转座体,因为转座体可以这样组装,通过将其转座酶的各个部分与相应的转座子dna结合,并且使2个转座酶二聚化以形成转座体,并且使转座体的2个转座子dna具有相同的条码序列。
根据一个方面,文库的各转座体成员包括在转座体的各转座子dna上相同序列的独特条码。以此方式,各转座体包括一对独特条码序列,其不同于转座体文库中任何其他转座体的条码序列。根据一个方面,转座体文库可以包括具有相同条码的转座体成员,尽管具有相同条码的成员数量相对较小或不显著。以此方式,可以认为转座体文库是制备的转座体集合的子集,其中该子集仅包括具有独特条码序列的转座体,因为目标是将基因组dna片段化,其中各片段切割位点由独特条码序列表示。应当理解的是,由于转座体文库制备,不显著数量的切割位点可能共有相同的条码序列。例如,对于给定的文库制备方法,数学上可能存在具有相同条码对的多个转座体分子,但是制备文库使得不同条码序列的数量将显著地超过实际将要插入靶基因组的转座体分子的数量。例如,对于6,000,000,000个碱基对长度的单个人细胞全基因组,需要将1,000,000个转座体插入全基因组以获得6,000bp的平均片段长度。为了实现该6000bp插入密度,将至少3,000,000,000个转座体分子添加到反应混合物中。对于14bp随机合成的条码,存在4^14=268,435,456个不同的条码序列,这意味着对于每个特异性条码,存在3,000,000,000/268,435,456=11.2个分子拷贝。但是无论多少分子拷贝共有相同的条码序列,具有相同条码序列的2个转座体分子插入基因组以产生片段的可能性是1,000,000/268,435,456=0.0037。藉由该示例,平均而言,在遇到具有相同条码标签或序列的2个不同的基因组dna片段之前,268个片段可通过条码连接。存在确保转座体文库中的各条码序列是唯一的方法,即以超过3,000,000,000个条码序列开始。
根据一个方面,对于其他大小的基因组,待使用的条码的数量可以相应缩放并且通过基因组中碱基对的总数量除以所需片段大小确定。例如,对于具有约50,000碱基对的小基因组,如λ噬菌体的小基因组,如果具有6,000bp的平均片段长度,仅需要9个条码插入基因组中,所以仅需要各自具有其独特的相关条码的9个转座体插入基因组。根据一个方面,通过使用更多或更少数量的转座体,平均片段长度还可以经调整地更小或更大,这可以通过分别使用更多或更少的浓缩转座体溶液实现;当目标平均片段长度较小以使得总片段的数量预期较大时,可以将待使用的条码或数量调整为更大以实现独特条码化,反之亦然。
因此,根据一个方面,基本上所有切割位点由独特条码序列表示,并且相应地,可以从头组装基本上所有片段。根据一个方面,超过90%的切割位点由独特条码序列表示,超过95%的切割位点由独特条码序列表示,96%的切割位点由独特条码序列表示,97%的切割位点由独特条码序列表示,98%的切割位点由独特条码序列表示,99%的切割位点由独特条码序列表示,99.5%的切割位点由独特条码序列表示或100%的切割位点由独特条码序列表示。
然后使用转座体文库切割基因组dna,并且各转座体将其条码序列(如独特条码序列)插入或连接位于切割位点两端的转座子dna中。以此方式,可以后续通过匹配条码序列鉴定切割位点的邻接末端,并且该邻接末端可以经计算连接在一起。根据一个方面,由转座体文库产生的片段在片段的各末端上具有条码序列对,如独特条码序列对的一个成员。根据一个方面,由转座体文库产生的片段在片段的各末端上各自具有条码序列对,如独特条码序列对的一个成员。在将片段扩增和测序后,可以通过匹配条码将片段的末端计算连接在一起,从而从头组装基因组dna。相应地,提供了用于通过匹配条码序列连接核酸片段的方法,所述条码序列已经使用转座酶与片段连接。
根据一个方面,转座体的转座子dna可以包括促进扩增方法的序列,如特异性引物序列或转录序列,其可以与片段连接,从而在诸如通过使用本领域技术人员已知的方法的pcr或rna转录进行测序之前,可以将片段扩增。应当理解的是,本公开考虑了用于扩增片段的不同的扩增方法以及用于对扩增子进行测序的不同的测序方法,并且用于基因组从头组装的方法并不限于任何特定的扩增或测序方法。
本公开的实施方式涉及dna的从头组装方法,如少量基因组dna或有限量dna,如获自单个细胞或相同细胞类型的多个细胞的一个或多个基因组序列或来自获自个体或底物的组织、液体或血液样品的一个或多个基因组序列。根据本公开的某些方面,本文所述的方法可以在具有单一反应混合物的单管中进行。根据本公开的某些方面,核酸样品可以在来自单个细胞的未纯化的或未处理的裂解物中。待进行本文所述方法的核酸,在将其与各种试剂接触并且经历本文所述的各种条件之前,不需要被纯化,如通过柱纯化。本文所述的条码方法辅助片段化的dna的从头组装,以便协助提供单个细胞整个基因组大量且均匀的覆盖,产生用于高通量测序的扩增dna。
本发明的实施方式通常涉及制造dna片段的方法和组合物,例如,来自单个细胞全基因组的dna片段,随后可对其进行本领域技术人员已知的以及本文所述的扩增和测序方法。根据某些方面,本文所述制造核酸片段的方法利用转座体文库根据一个方面,作为转座体一部分的转座酶用于产生一组双链基因组dna片段。根据某些方面,转座酶具有结合转座子dna以及当接触时(如将其置于反应容器或反应体积内时)二聚化,形成转座酶/转座子dna复合物二聚体的能力,该转座酶/转座子dna复合物二聚体被称为转座体。转座体的各转座子dna包含双链转座酶结合位点和第一核酸序列,其包含对转座体独特的条码序列和扩增促进序列,如特异性引物位点(“引物结合位点”)或转录启动子位点。第一核酸序列可以处于单链延伸的形式。转座体文库的各转座体包括独特条码序列,其不同于转座体文库各剩余成员的条码序列。
转座体具有随机结合沿双链核酸(如双链基因组dna)分布的靶位置形成包括转座体和双链基因组dna的复合物的能力。转座体中的转座酶切割双链基因组dna,其中一个转座酶切割上链,一个转座酶切割下链。转座体中各转座子dna在切割位点的各末端与双链基因组dna连接,即转座体的一个转座子dna与左手切割位点连接,而转座体的另一转座子dna与右手切割位点连接。以此方式,用对切割位点独特的相同的条码序列将左手切割位点和右手切割位点条码化。相应地,条码序列鉴定左手切割位点和右手切割位点为彼此直接邻接用于基因组从头组装。
根据某些方面,例如,多个转座酶/转座子dna复合物二聚体(即,转座体)结合沿双链基因组dna分布的相应的多个靶位置,并且然后将双链基因组dna切割成多个双链片段,其中各片段具有不同条码序列的转座子dna,所述条码序列连接于双链片段各末端。以此方式并与上述描述相一致,通过鉴定具有相同条码序列的片段的相应末端并且将片段的末端计算连接在一起,可以将各片段计算放置于序列中。
根据一个方面,转座子dna连接于双链基因组dna,并且单链缺口存在于基因组dna的一条链和转座子dna的一条链之间。根据一个方面,进行缺口延伸以填平缺口并产生双链基因组dna和双链转座子dna之间的双链连接。根据一个方面,包含转座子dna的扩增促进序列、转座酶结合位点、和条码序列的核酸序列连接于双链片段的各末端。根据某些方面,转座酶连接转座子dna,所述转座子dna连接于双链片段的各末端。根据一个方面,将转座酶从转座子dna去除,所述转座子dna连接于双链基因组dna片段的各末端。
根据本发明的一个方面,然后用转座子dna作为模板对通过转座酶产生的双链基因组dna片段进行缺口填平和延伸,所述转座酶具有含有不同条码序列的转座子dna,所述条码序列连接于双链基因组dna片段的各末端。相应地,产生双链核酸延伸产物,其包括双链基因组dna片段和位于双链基因组dna各末端的双链转座子dna,其包含不同的条码序列和扩增促进序列。
在该阶段,可以使用本领域技术人员已知的方法扩增双链核酸延伸产物以产生位于各末端的不同的条码和基因组dna片段的扩增子,所述双链核酸延伸产物包含基因组dna片段,位于各末端的不同的条码和扩增促进序列。扩增促进序列可以是位于双链基因组dna各末端的特异性引物结合位点。所述“特异性”引物结合位点指示2个引物结合位点具有相同序列,并且因此共同序列的引物可以用于扩增所有片段。pcr引物序列和试剂可以用于扩增。扩增促进序列可以是用于产生rna转录本的rna聚合酶结合位点,所述rna转录本然后可以被逆转录成cdna用于线性扩增。使用本领域技术人员已知的方法,双链核酸延伸产物可以与扩增试剂合并,然后可以扩增双链基因组核酸片段,以产生双链基因组核酸片段的扩增子,所述双链核酸延伸产物包含基因组dna片段,位于各末端的不同的条码和扩增促进序列。
然后在进一步分析之前可以收集和/或纯化扩增子。可使用本领域技术人员已知的方法对扩增子进行测序。一旦测序,可以计算分析序列以鉴定具有相同条码序列的片段末端,并且可以彼此计算连接片段末端以产生用于基因组dna从头组装的较长序列。在一实施方式中,当基因组dna来自具有超过1个倍性的单个细胞时,基因组的从头组装可以实现单体型分辨率的从头组装,当独特条码序列被插入2个等位基因各自片段的各片段末端时。
本公开的实施方式涉及使用本文所述的条码化片段扩增dna的方法,其中dna是少量基因组dna或有限量dna,如获自单个细胞或相同细胞类型的多个细胞的一个或多个基因组序列或来自获自个体或底物的组织、液体或血液样品的一个或多个基因组序列。根据本公开某些方面,本文所述方法可以在单管中进行以产生条码化的片段,然后使用本领域技术人员已知的高通量测序平台将其扩增并测序,并且然后使用本领域技术人员已知的方法和软件,通过匹配条码序列将末端与末端计算连接,所述条码序列指定原始核酸序列的邻接片段之间的切割或片段化位点。
本文所述的转座体片段化和条码化方法能够用于扩增、测序和从头组装少量或有限量的dna。本文所述方法在以高异质细胞群(如肿瘤和神经快)为特征的组织样品或生物系统中具有特定应用。本文所述扩增并测序条码化基因组dna片段的方法促进使用本领域技术人员已知以及本文所述的下一代测序技术对这样扩增的dna的分析和从头组织。本文所述的方法可以利用不同来源的dna材料,包括遗传异质性组织(例如,癌症),稀有和珍贵样品(例如,胚胎干细胞),和非分裂细胞(例如,神经元)等,以及本领域技术人员已知的测序平台和基因分型方法。
本公开的某些实施方式的其他特征和优势将在权利要求中以及以下附图和实施方式的说明下更为显而易见。
附图说明
结合附图,通过以下示例性实施方式的详述能够更清楚地理解本发明实施方式的上述和其他特征和其他优点,其中:
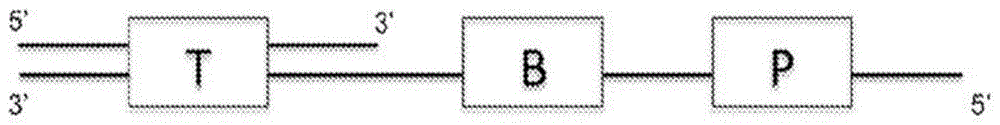
图1在示意图中描述了具有线性5'延伸的转座子dna的结构,其中t是双链转座酶结合位点,p是位于延伸一侧的引发位点,而b是条码序列。
图2是转座酶和转座子dna自发地形成转座体的一般实施方式的示意图,其可以在液滴或其他形成培养基中出现。
图3是这样的转座体的示意图,所述转座体结合基因组dna,切割成片段并且添加或插入包含引物结合位点(紫色)、转座酶结合位点(浅蓝色)和在各转座体中通过不同颜色表示的独特条码序列的转座子dna。
图4是转座酶去除、缺口填平和延伸以形成核酸延伸产物的示意图,所述核酸延伸产物包含基因组dna、引物结合位点、条码序列和转座酶结合位点。
图5是使用条码将短的测序读数栓链(chain)到较长连续序列中的示意图。
图6描述了这样的微粒或珠,其具有通过接头与其连接的多个转座子dna并且具有用于从微粒或珠切割转座子dna的切割位点。
图7是使用微滴来分离含有具有特异性条码的转座子dna的微粒,以及在各微滴内产生具有相同条码对的转座体的示意图。
图8是用于制备条码化的转座体的微流体回路的示意图。
图9是将携带不同条码对的转座体插入二倍体基因组的2个等位基因以及单体型分析(haplotype)基因组的示意图。
具体实施方式
除非另有说明,某些实施方式的实践或某些实施方式的特征可以采用分子生物学、微生物学、重组dna和免疫学和本技术领域内的常规技术。这些技术在文献中已有充分描述。参见,例如,sambrook,fritsch,和maniatis,《分子克隆:实验室手册(molecularcloning:alaboratorymanual)》,第二版(1989),《寡核苷酸合成(oligonucleotidesynthesis)》(m.j.gait编著,1984),《动物细胞培养(animalcellculture)》(r.i.freshney编著,1987),《酶学方法(methodsinenzymology)》丛书(学术出版社有限公司(academicpress,inc.));《哺乳动物细胞的基因转移载体(genetransfervectorsformammaliancells)》(j.m.miller和m.p.calos编著.1987),《免疫学实验手册(handbookofexperimentalimmunology)》,(d.m.weir和c.c.blackwell编著),《新编分子生物学实验指南(currentprotocolsinmolecularbiology)》(f.m.ausubel,r.brent,r.e.kingston,d.d.moore,j.g.siedman,j.a.smith,和k.struhl编著,1987),《新编免疫学实验指南(currentprotocolsinimmunology)》(j.e.coligan,a.m.kruisbeek,d.h.margulies,e.m.shevach和w.strober编著,1991);《免疫学年鉴(annualreviewofimmunology)》;以及如《免疫学进展(advancesinimmunology)》等期刊中的专著。本文上下文中提及的所有专利、专利申请和出版物均以参考的方式用全文纳入本文。
本文所用的核酸化学、生物化学、遗传学和分子生物学的术语和符号遵循本领域的标准论述和文本中的术语和符合,例如,kornberg和baker,dnareplication(《dna复制》),第二版(w.h.弗里曼出版社(w.h.freeman),纽约,1992);lehninger,biochemistry(《生物化学》),第二版(沃斯出版社(worthpublishers),纽约,1975);strachan和read,humanmoleculargenetics(《人类分子遗传学》),第二版(wl出版社(wiley-liss),纽约,1999);eckstein编,oligonucleotidesandanalogs:apracticalapproach(《寡核苷酸和类似物:实践方法》)(牛津大学出版社(oxforduniversitypress),纽约,1991);gait编,oligonucleotidesynthesis:apracticalapproach(《寡核苷酸合成:实践方法》)(irl出版社,牛津,1984);等。
本发明部分基于用于制造核酸片段模板的方法的发现,如从dna或基因组dna,使用转座酶或转座体使原始或起始核酸序列(如基因组dna)片段化,并且将条码序列连接于切割或片段化位点的各末端以促进作为从头组装过程一部分的片段序列的后续计算再连接。本文所述方法可以称之为“经由转座子插入的链注释”或“chianti”。扩增条码化的核酸片段模板以产生扩增子。可以收集并对核酸片段模板的扩增子进行测序。收集的扩增子形成原始核酸(如基因组dna)片段扩增子的文库。
根据一个方面,获得基因组dna,如获自裂解的单个细胞的基因组核酸。使用多个转座体或转座体文库将基因组dna切割成双链片段。多个转座体或转座体文库中的每一个转座体是结合转座子dna的转座酶的二聚体,即,各转座体包含2个单独的转座子dna。转座体的各转座子dna包含转座酶结合位点,对转座体独特的条码序列和扩增促进序列,如特异性引物结合位点。
转座体的各转座子dna的条码序列是相同的序列并且对转座体是独特的。多个转座体或转座体文库中的每一个转座体具有其独特的代表性条码序列,其不同于多个转座体或转座体文库中剩余的成员。转座体dna在各切割或片段化位点连接各双链片段的上链和下链。因为条码序列对于各转座子dna是相同的,所以用相同的条码序列标签化切割或片段化位点,所述条码序列随后可以经鉴定以计算再连接切割或片段化位点。因为各转座体具有其自身独特的条码序列,并且转座体文库被用于产生许多切割或片段化位点,各切割或片段化位点将具有其自身独特的条码序列。相应地,来自原始核酸序列的许多片段由转座体的文库产生,其中各片段在片段的每个末端具有不同的条码。然后处理双链片段以填平缺口。使用合适的扩增试剂,如用于pcr扩增的核苷酸,特异性引物序列和dna聚合酶,扩增片段并使用本领域技术人员已知的方法进行测序。鉴定匹配的条码,其指示切割或片段化位点,并且将匹配的条码用于计算再连接片段以再生原始核酸序列。
使用本文所述方法制造的dna片段模板可以使用本领域技术人员已知的方法在微滴内进行扩增。微滴可以作为油相或水相的乳液形成。乳液可以包括连续油相内分离的水性体积或水性液滴。描述了乳液全基因组扩增方法,使用油内的小体积水性液滴来分离各片段,用于单个细胞基因组的统一扩增。通过将各片段分配到其自身的液滴或分离的水性反应体积,允许各液滴达到dna扩增的饱和。然后,将各液滴内的扩增子通过破乳作用合并,产生单个细胞全基因组所有片段的均一扩增。
在某些方面,扩增使用pcr实现。pcr是这样一种反应,其中使用由上游和下游引物组成的一组引物或一对引物和聚合催化剂(如dna聚合酶)以及热稳定的聚合酶(通常使用),由靶多核苷酸制备复制拷贝。pcr的方法在本领域是公知的,并且在例如macpherson等.(1991)pcr1:使用方法(pcr1:apracticalapproach)牛津大学出版社(oxforduniversitypress)irl出版社(irlpress)中教导。mullis(美国专利号4,683,195、4,683,202和4,965,188)的术语“聚合酶链反应”(“pcr”)指用于在未克隆或纯化的情况下提高靶序列区段浓度的方法。用于扩增靶序列的方法包括提供具有所需靶序列的寡核苷酸引物和扩增试剂,然后在聚合酶(例如,dna聚合酶)存在的情况下进行准确的一连串热循环。引物与双链靶序列各自的链(“引物结合序列”)互补。为了进行扩增,将双链靶序列变性,然后将引物退火至靶分子中的其互补序列。退火后,用聚合酶延伸引物,从而形成一对新的互补链。变性、引物退火和聚合酶延伸步骤可以重复多次(即,变性、退火或延伸组成一个“循环”;可以存在许多“循环”)以获得所需靶序列的高浓度扩增区段。所述靶序列的扩增区段的长度由引物相对于彼此的相对位置确定,并且因此,长度是可控参数。由于该过程的重复,该方法被称为“聚合酶链反应”(下文称之为“pcr”)并且靶序列被称为是“pcr扩增的”。当双链dna扩增产物累积到dna聚合酶活性被抑制的一定量时,pcr扩增达到饱和。一旦饱和,pcr扩增达到稳定水平,此时扩增产物不会随着pcr循环的增加而增加。
藉由pcr,有可能将基因组dna中的特定靶序列的单拷贝扩增至通过几种不同方法(例如,与标记探针杂交;纳入生物素化引物,然后进行抗生物素蛋白-酶偶联物检测;将32p-标记的脱氧核苷酸三磷酸(如dctp或datp)纳入扩增区段)可检测的水平。除了基因组dna,任何寡核苷酸或多核苷酸都可以用适当的引物分子组进行扩增。特别是,在各微滴内通过pcr过程其自身产生的扩增区段本身就是用于后续pcr扩增的有效模板。用于进行pcr的方法和试剂盒是本技术领域已知的。产生多核苷酸复制拷贝的所有方法(如pcr或基因克隆)在本文中统称为复制。引物还可以用作杂交反应中的探针,如southern或northern印迹分析。
“扩增”或“进行扩增”这样的表达指通过通过其将形成特定多核苷酸的额外或多个拷贝的过程。扩增包括诸如pcr、连接扩增(或连接酶链反应,lcr)和其他扩增方法的方法。这些方法在本领域中是已知且广泛应用的。参见,例如,美国专利号4,683,195和4,683,202,以及innis等.,"pcr方法:方法和应用的指南(pcrprotocols:aguidetomethodandapplications)'”学术出版社股份有限公司公司(academicpress,incorporated)(1990)(针对pcr);和wu等.(1989)genomics4:560-569(针对lcr)。通常,pcr过程描述了这样一种基因扩增方法,其包括(i)引物与dna样品(或文库)中特定基因的序列特异性杂交,(ii)随后的扩增,涉及使用dna聚合酶地多轮退火、延伸和变性,和(iii)筛选pcr产物以获得正确大小的条带。使用的引物是具有足够长度和适当序列的寡核苷酸以引发聚合,即特异性地涉及各引物,使其与待扩增的基因组基因座的每条链互补。
进行扩增反应的试剂和硬件是市售可得的。用于从特定基因区域扩增序列的引物优选与目标区域或其侧接区中的序列互补并与其特异性杂交,并且可以使用本领域技术人员已知道的方法制备。通过扩增生成的核酸序列可以直接进行测序。
当杂交以两个单链多核苷酸之间的反平行构型发生时,该反应被称为“退火”,并且这些多核苷酸被描述为“互补的”。如果杂交可以发生在第一多核苷酸的一条链与第二多核苷酸的链之间,那么双链多核苷酸可以与另一多核苷酸互补或同源。根据普遍接受的碱基配对规则,互补性或同源性(一个多核苷酸与另一个多核苷酸互补的程度)可依据相对链中预计将彼此之间形成氢键的碱基的比例来定量。
术语“pcr产物”、“pcr片段”和“扩增产物”指在变性、退火和延伸pcr步骤的两个或更多个循环完成后得到的化合物混合物。这些术语包括已经扩增了一个或多个靶序列的一个或多个片段的情况。根据本公开的一个方面,各微滴包含单个模板dna片段的pcr产物。
术语“扩增试剂”可以指除了引物、核酸模板和扩增酶以外扩增所需的那些试剂(脱氧核糖核苷三磷酸,缓冲液等)。通常,将扩增试剂与其他反应组分一起放置并容纳在反应容器中(试管,微孔等)。扩增方法包括本领域技术人员已知的pcr方法,并且还包括滚环扩增(blanco等.,j.biol.chem.,264,8935-8940,1989)、超支化滚环扩增(lizard等.,nat.genetics,19,225-232,1998)和环介导的等温扩增(notomi等.,nuc.acidsres.,28,e63,2000),其各自通过引用将其全部内容纳入本文。
对于乳液pcr,通过剧烈振荡或搅拌“油包水”混合物以生成数百万个微米级水性隔室来产生乳液pcr反应。设备可以装备微流体芯片以通过摇晃或搅拌油相和水相产生乳液。或者,通过将某些油与水相合并或通过将水相导入油相可以自发地形成水性液滴。待扩增的dna文库在乳化之前以有限稀释混合。隔室大小(即微滴尺寸)以及产生待扩增dna片段文库有限稀释的微滴数量的组合被用于生成平均只包含一个dna分子的隔室。基于微滴形成或乳化步骤中生成的水性隔室的大小,可以在同一管中同时进行每μl上至3x109个单独的pcr反应。基本上乳液中的各小水性隔室微滴形成微型pcr反应器。乳液中隔室的平均尺寸根据乳化的条件范围从亚微米直径至超过100微米,或从1皮升(picoliter)至1000皮升,或从1纳升至1000纳升,或从1皮升至1纳升,或从1皮升至1000纳升。
其他扩增方法,如英国专利申请号gb2,202,328以及pct专利申请号pct/us89/01025中所述的方法,各自通过引用纳入本文,可以依据本公开使用。在前一申请中,“修饰的”引物被用于pcr样模板和酶依赖性合成中。引物可以通过用捕获部分(例如,生物素)和/或检测器部分(例如,酶)标记来修饰。在后一申请中,将过量的标记探针添加到样品。在靶序列存在的情况中,探针结合并且被催化切割。切割后,靶序列被完整的释放,以被过量的探针结合。标记探针的切割表面靶序列的存在。
其它合适的扩增方法包括“race”和“单侧pcr”(frohman,述于《pcr方案:方法和引用的指南(pcrprotocols:aguidetomethodsandapplications)》,学术出版社,纽约,1990,其各自通过引用纳入本文)。基于在具有所得“二寡核苷酸”序列的核酸存在的情况下连接两个(或多个)寡核苷酸并因此扩增该二寡核苷酸的方法也可以用于根据本公开扩增dna(wu等.,genomics4:560-569,1989,通过引用纳入本文)。
根据某些方面,示例性的转座子系统包含tn5转座酶,mu转座酶,tn7转座酶或is5转座酶等。其它可用的转座子系统是本领域技术人员已知的,并且包括tn3转座子系统(参见maekawa,t.,yanagihara,k.,和ohtsubo,e.(1996),tn3转座的无细胞系统和转座免疫(acell-freesystemoftn3transpositionandtranspositionimmunity),genescells1,1007-1016)、tn7转座子系统(参见craig,n.l.(1991),tn7:靶向位点特异性转座子(tn7:atargetsite-specifictransposon),mol.microbiol.5,2569-2573)、tn10转座子系统(参见chalmers,r.,sewitz,s.,lipkow,k.,和crellin,p.(2000),tn10的完整核苷酸序列(completenucleotidesequenceoftn10),j.bacteriol182,2970-2972)、piggybac转座子系统(参见li,x.,burnight,e.r.,cooney,a.l.,malani,n.,brady,t.,sander,j.d.,staber,j.,wheelan,s.j.,joung,j.k.,mccray,p.b.,jr.,等.(2013),用于基因工程的piggybac转座酶工具(piggybactransposasetoolsforgenomeengineering),proc.natl.acad.sci.usa110,e2279-2287)、睡美人转座子系统(参见ivics,z.,hackett,p.b.,plasterk,r.h.,和izsvak,z.(1997),来自鱼类的睡美人、tcl样转座子的分子重建及其在人细胞中的转座(molecularreconstructionofsleepingbeauty,atc1-liketransposonfromfish,anditstranspositioninhumancells),cell91,501-510)、tol2转座子系统(参见kawakami,k.(2007),tol2:脊椎动物中多功能基因转移载体(tol2:aversatilegenetransfervectorinvertebrates),genomebiol.8增刊.1,s7.)。
待扩增的dna可以获得自单个细胞或小细胞群。本文所述方法允许由反应混合物中的任何物种或生物体扩增dna,如在单个反应容器中进行的单一反应混合物。在一个方面中,本文所述方法包括由任何来源进行dna的序列非依赖性扩增,所述来源包括但不限于人、动物、植物、酵母、病毒、真核和原核dna。
根据一个方面,提供了单细胞全基因组扩增、测序和从头组装的方法,其包括使来自单个细胞的双链基因组dna与各自结合转座子dna的tn5转座酶接触,其中转座子dna包括双链19bp的转座酶(tnp)结合位点和第一核酸序列以形成被称之为转座体的转座酶/转座子dna复合物二聚体,所述第一核酸序列包括条形码序列和引物结合位点中的一个或多个。第一核酸序列可以处于单链延伸的形式。根据一个方面,第一核酸序列可以是突出端,如5’突出端,其中该突出端包含条码区域和引发位点。该突出端可以是适合包含所需条码区域和引发位点的任何长度。转座体沿着双链基因组dna结合靶位置并将双链基因组dna切割成多个双链片段,各双链片段具有通过tnp结合位点连接上链的第一复合物,以及通过tnp结合位点连接下链的第二复合物。转座子结合位点以及因此的转座子dna连接双链片段各5’末端。根据一个方面,将tn5转座酶从复合物去除。沿着转座子dna延伸双链片段以制造这样的双链延伸产物,其在双链延伸产物的各末端具有不同的条码序列和特异性引物结合位点。根据一个方面,可能是由于tn5转座酶结合位点与双链基因组dna片段所导致的缺口可以被填充。经缺口填平的双链延伸产物与扩增试剂混合,并将双链基因组dna片段扩增。使用例如本领域技术人员已知的高通量测序方法对扩增子进行测序,所述扩增子在各末端包含不同的条码序列。
在一个具体方面中,实施方式涉及用于在不丧失特异性位点的表现度的情况下扩增、测序和从头组装基本上整个基因组的方法(本文定义为“全基因组扩增”)。在特定实施方式中,全基因组扩增包括同时扩增基因组文库基本上所有片段或所有片段。在另一个特定实施方式中,“基本上整个”或“基本上所有”指基因组中约80%、约85%、约90%、约95%、约97%、或约99%的所有序列。
根据一个方面,dna样品是基因组dna,显微解剖的染色体dna,酵母人工染色体(yac)dna,质粒dna,粘粒dna,噬菌体dna,p1衍生的人工染色体(pac)dna或细菌人工染色体(bac)dna,线粒体dna,叶绿体dna、司法鉴定样品dna或来自待测试的自然或人工来源的其他dna。在另一优选实施方式中,dna样品是哺乳动物dna、植物dna、酵母dna、病毒dna或原核生物dna。在又一优选实施方案中,dna样品获自人、牛、猪、羊、马、啮齿动物、禽、鱼、虾、植物、酵母、病毒或细菌。优选地,dna样品是基因组dna。
根据某些示例性方面,转座系统被用于制造用于如所需进行扩增、测序和从头组装的核酸片段。根据一个方面,转座系统被用于将基因组dna片段化成双链基因组dna片段,其中转座子dna具有插入其中的相同条码。如图1所示,转座子dna包含双链转座酶结合位点、条码序列b和引发位点p。双链转座酶结合位点可以是双链19bp的tn5转座酶(tnp)结合位点,其诸如通过共价键连接或接合单链突出端,其包含位于突出端一个末端的引发位点和条码区。将转座子dna插入单个细胞的基因组dna,同时使用转座酶产生数百万个小片段。转座酶去除和缺口填平后,使用特异性引物以及dna聚合酶、核苷酸和扩增试剂扩增基因组dna片段,以pcr扩增单细胞的全基因组,所述基因组dna片段具有位于片段各末端的特异性引物和不同的条码序列。
根据某些方面,当扩增少量dna(如来自单细胞的dna)时,不进行dna柱纯化步骤,从而使可以在扩增前从单细胞内获得的少量(约6pg)基因组dna最大化。dna可以从细胞裂解物或其它不存条件直接扩增。相应地,dna样品可以是不纯的、未纯化的或未分离的。相应地,本方法的方面允许人们使用于扩增的基因组dna最大并且减少由于纯化的损失。根据其它方面,本文所述方法可以利用除了pcr以外的扩增方法。
根据一个方面并且如图2中所示,将转座酶(tnp)和转座子dna合并,如在微滴内,并且tnp和转座子dna彼此结合并且二聚化以形成转座体。
如图3所示,转座体文库的转座体随机捕获或以其它方式与靶向单个细胞基因组dna结合成二聚体。代表性的转座体编号为1、2和3,虽然转座体数可以是数千个,数万个,数十万个,数百万个等。各转座体由独特条码序列表示,例如,条码序列1、条码序列2、条码序列3等。独特条码序列在转座体的各转座子dna内。因为每个转座体存在2个转座子dna,可以将这2个转座子dna认为是同源二聚体,这意味着1个转座子dna二聚体携带具有相同条码信息的2个dna序列。转座体文库的各转座体(和转座子dna二聚体)具有独特于转座体的不同的条码。转座体中的转座酶切割基因组dna,其中一个转座酶切割上链而一个转座酶切割下链以产生基因组dna片段。多个转座体产生多个基因组dna片段。因此,来自转座体dna二聚体的一个转座子dna连接切割位点或片段化位点的各末端,即来自转座体1的一个转座子dna与左手切割位点连接,而来自转座体1的另一转座子dna与右手切割位点连接。因为转座体文库将核酸切割成片段,各片段在片段的各末端将具有不同的条码序列,即通过2个不同的切割位点产生各片段,所述2个不同的切割位点由包含不同条码序列的转座体文库的2个不同转座体切割。这通过2个示例性片段表示,其中上片段在一末端具有条码序列1并且在另一末端具有条码序列2。同样,下片段在以末端具有条码序列2并且在另一末端具有条码序列3。如所示,通过转座体2和左手切割位点(即,观看图3中上片段的右侧)产生的2个片段之间的切割位点包含具有条码序列2的一个转座子,而右手切割位点(即,观看图3中下片段的左侧)包含具有条码序列2的另一个转座子。
如图4所示,基因组dna的片段化在转座/插入位点的末端留下缺口。缺口可能是任何长度,但是示例性的是9个碱基缺口。结果是这样的基因组dna片段,其具有连接上链的5'位置的转座子dnatnp结合位点以及连接下链的5'位置的转座子dnatnp结合位点。示出了由于连接或插入转座子dna而产生的缺口。在转座后,转座酶被去除并且进行缺口延伸以填平缺口并且与最初设计在如图4所示的转座子dna中的单链突出端互补。
如图5进一步所示,具有相应条码序列bn的多个转座体n用于产生多个片段,而条码序列用于将短测序读数栓链到较长连续序列中。将具有这样转座体的转座体文库(例如,以百万计)插入基因组dna并切割基因组dna成数百万不同的片段(f1、f2、f3……),所述转座体各自携带具有相同条码b(n)的2个转座子dna。全基因组扩增和测序后,用相同条码标签标记的片段可以计算连接在一起以实现较长的片段长度。
描述了特定的tn5转座子系统,并且是本领域技术人员可使用的。参见goryshin,i.y.和w.s.reznikoff,tn5体外转座(tn5invitrotransposition).thejournalofbiologicalchemistry,1998.273(13):7367-74页;davies,d.r.,等.,tn5突触复合物转座中间体的三维结构(three-dimensionalstructureofthetn5synapticcomplextranspositionintermediate).science,2000.289(5476):77-85页;goryshin,i.y.,等.,通过电穿孔释放的tn5转座复合物的插入转座子突变(insertionaltransposonmutagenesisbyelectroporationofreleasedtn5transpositioncomplexes).naturebiotechnology,2000.18(1):97-100页,以及steiniger-white,m.,i.rayment,和w.s.reznikoff,tn5转座的结构/功能研究(structure/functioninsightsintotn5transposition).currentopinioninstructuralbiology,2004.14(1):50-7页,其各自通过引用出于所有目的将其全部内容纳入本文。利用tn5转座系统进行dna文库制备和其它应用的试剂盒是已知的。参见adey,a.,等.,通过高密度体外转座来快速、低输入、低偏倚构建鸟枪片段文库(rapid,low-input,low-biasconstructionofshotgunfragmentlibrariesbyhigh-densityinvitrotransposition).genomebiology,2010.11(12):r119页;marine,r.,等.,评估用于从纳克量的dna快速生成鸟枪高通量测序文库的转座酶方案(evaluationofatransposaseprotocolforrapidgenerationofshotgunhigh-throughputsequencinglibrariesfromnanogramquantitiesofdna).appliedandenvironmentalmicrobiology,2011.77(22):8071-9页;parkinson,n.j.,等.,由微微克量的靶dna制备高质量下一代测序文库(preparationofhigh-qualitynext-generationsequencinglibrariesfrompicogramquantitiesoftargetdna).genomeresearch,2012.22(1):125-33页;adey,a.和j.shendure,超低输入、基于标签作用的全基因组亚硫酸氢盐测序(ultra-low-input,tagmentation-basedwhole-genomebisulfitesequencing).genomeresearch,2012.22(6):1139-43页;picelli,s.,等.,使用smart-seq2由单个细胞的全长rna-seq(full-lengthrna-seqfromsinglecellsusingsmart-seq2).natureprotocols,2014.9(1):171-81页,以及buenrostro,j.d.,等.,天然染色质的转座用于开放染色质、dna结合蛋白和核小体位置的快速和敏感表观基因组概况(transpositionofnativechromatinforfastandsensitiveepigenomicprofilingofopenchromatin,dna-bindingproteinsandnucleosomeposition).naturemethods,2013,其各自通过引用出于所有目的将其全部内容纳入本文。同样参见wo98/10077、ep2527438和ep2376517,其各自通过引用出于所有目的将其全部内容纳入本文。市售可得的转座试剂盒以nextera的名称销售并可从illumina公司获得。
本文所用术语“基因组”被定义为由个体、细胞或细胞器携带的总体基因(collectivegene)集合。本文所用术语“基因组dna”被定义为这样的dna材料,其包含由个体、细胞或细胞器携带的部分或全部集体基因集合。
本文所用术语“核苷”是指具有与核糖或脱氧核糖共价连接的嘌呤或嘧啶碱基的分子。示例性的核苷包括腺苷、鸟苷、胞苷、尿苷和胸苷。其它示例性的核苷包括肌苷、1-甲基肌苷、假尿苷、5,6-二氢尿苷、核糖胸核苷、2n-甲基鸟苷和2,2n,n-二甲基鸟苷(也称为“稀有”核苷)。术语“核苷酸”是指具有一个或多个与糖部分以酯键连接的磷酸基团的核苷。示例性的核苷酸包括核苷单磷酸酯、二磷酸酯和三磷酸酯。术语“多核苷酸”、“寡核苷酸”和“核酸分子”在本文中可互换使用,并指通过5'和3'碳原子之间的磷酸二酯键连接在一起的任何长度的核苷酸(脱氧核糖核苷酸或核糖核苷酸)的聚合物。多核苷酸可以具有任何三维结构并且可以进行已知或未知的任何功能。以下是多核苷酸的非限制性例子:基因或基因片段(例如,探针、引物、est或sage标签)、外显子、内含子、信使rna(mrna)、转移rna、核糖体rna、核酶、cdna、重组多核苷酸、支链多核苷酸、质粒、载体、任意序列的分离dna、任意序列的分离rna、核酸探针和引物。多核苷酸可以包括修饰的核苷酸,如甲基化的核苷酸和核苷酸类似物。该术语也指双链和单链分子。除非另有说明或要求,本发明包含多核苷酸的任何实施方式都包括双链形式和已知或预测构成双链形式的两种互补单链形式中的每一种。多苷酸由4个核苷酸碱基的特定序列组成:腺嘌呤(a);胞嘧啶(c);鸟嘌呤(g);胸腺嘧啶(t);并且当多核苷酸是rna时,尿嘧啶(u)替代胸腺嘧啶(t)。因此,术语多核苷酸序列是多核苷酸分子的字母表示。可以将该字母表示输入具有中央处理单元的计算机中的数据库中,并用于生物信息学应用,如功能基因组学和同源性搜索。
术语“dna”、“dna分子”和“脱氧核糖核酸分子”指脱氧核糖核苷酸的聚合物。可以自然合成dna(例如,通过dna复制)。可以对rna进行转录后修饰。也可以化学合成dna。dna可以是单链(即ssdna)或多链(例如,双链,即dsdna)。
术语“核苷酸类似物”、“改变的核苷酸”和“修饰的核苷酸”指非标准核苷酸,包括非天然存在的核糖核苷酸或脱氧核糖核苷酸。在某些示例性实施方式中,在任何位置修饰核苷酸类似物,从而改变核苷酸的某些化学性质,但仍保留核苷酸类似物进行其预期功能的能力。可被衍生化的核苷酸位置的示例包括5位置,例如,5-(2-氨基)丙基尿苷、5-溴尿苷、5-丙炔尿苷,5-丙烯基尿苷等;6位置,例如,6-(2-氨基)丙基尿苷:腺苷和/或鸟苷的8-位,例如,8-溴鸟苷,8-氯鸟苷,8-氟鸟苷等。核苷酸类似物还包括脱氮核苷酸,例如,7-脱氮腺苷;o-和n-修饰的(例如,烷基化的,例如,n6-甲基腺苷,或如本领域其他已知的)核苷酸;以及其他杂环修饰的核苷酸类似物,如herdewijn,antisensenucleicaciddrugdev.,2000年8月10日(4):297-310中所述的那些。
核苷酸类似物还可以包括对于核苷酸糖部分的修饰。例如,2'oh-基团可被选择如下的基团取代:h、or、r、f、cl、br、i、sh、sr、nh2、nhr、nr2、coor、或or,其中,r是取代的或未取代的c1-c6烷基、链烯基、炔基、芳基等。其它可能的修饰包括在美国专利号5,858,988,和6,291,438中所述的那些。
也可对核苷酸的磷酸基团进行修饰,例如,通过用硫取代磷酸基团的一个或多个氧(例如,硫代磷酸酯),或通过进行允许核苷酸发挥其预期功能的其他取代方式,如在例如eckstein,antisensenucleicaciddrugdev.2000年4月10日(2):117-21、rusckowski等.antisensenucleicaciddrugdev.2000年10月10日(5):333-45、stein,antisensenucleicaciddrugdev.2001年10月11日(5):317-25、vorobjev等.antisensenucleicaciddrugdev.2001年4月11日(2):77-85和美国专利号5,684,143中所述。例如,某些上述修饰(例如,磷酸基团修饰)降低了体内或体外包含所示类似物的多核苷酸的水解速率。
术语“体外”具有其本技术领域公认的含义,例如,涉及纯化的试剂或提取物,例如,细胞提取物。术语“体内”还具有其本技术领域公认的含义,例如,涉及活细胞,例如,生物体中的永生化细胞、原代细胞、细胞系和/或细胞。
本文所用术语“互补”和“互补性”用于指通过碱基配对规则相关联的核苷酸序列。例如,序列5'-agt-3'与序列5'-act-3'互补。互补性可以是部分的或完全的。部分互补性发生在当一个或多个核酸碱基根据碱基配对规则不匹配时。核酸间完全或完整互补性发生在各和每个核酸碱基在碱基配对规则下与另一个碱基匹配时。核酸链间的互补性程度对于核酸链间杂交的效率和强度有显著影响。
术语“杂交”是指互补核酸的配对。杂交和杂交的强度(即核酸之间关联的强度)受诸如核酸之间的互补性程度,涉及条件的严谨性,形成的杂交体的tm和核酸内g:c比例的影响。在其结构中包含互补核酸配对的单个分子是“自交的”。
术语“tm”指核酸的解链温度。解链温度是双链核酸分子群体一半解离成单链的温度。计算核酸tm的等式是本领域熟知的。如标准参考文献所示,当核酸处于1mnacl水性溶液中时,通过tm=81.5+0.41(%g+c)等式可以简单估计tm值(参见,例如,anderson和young,定量滤膜杂交(quantitativefilterhybridization),innucleicacidhybridization(1985))。其他参考文献包括更复杂的计算,它们将结构以及序列特性考虑到tm的计算中。
术语“严谨性”指进行核酸杂交的温度,离子强度和存在其他化合物(如有机溶剂)的条件。
当就核酸杂交使用时,“低严谨性条件”包括等同于在42℃这样的溶液中结合或杂交的条件,所述溶液由5xsspe(43.8g/lnacl、6.9g/lnah2po4(h2o)和1.85g/ledta,用naoh将ph调至7.4with)、0.1%sds、5xdenhardt试剂(50xdenhardt试剂每500ml含:5gficoll(400型,法玛西亚公司(pharmacia))、5gbsa(组分v;西格玛公司(sigma)))和100mg//ml变性的鲑鱼精dna组成,然后当使用约500核苷酸长度探针时,在42℃包括5xsspe、0.1%sds的溶液中洗涤。
当述及核酸杂交时,所用的“中严谨性条件”包括等同于在42℃这样的溶液中结合和杂交的条件,所述溶液由5xsspe(43.8g/lnacl、6.9g/lnah2po4(h2o)和1.85g/ledta、用naoh将ph调节至7.4)、0.5%sds、5xdenhardt试剂和100mg/ml变性的鲑鱼精组成,然后当使用约500核苷酸长度的探针时,在42℃包括1.0xsspe、1.0%sds的溶液中洗涤。
当述及核酸杂交时,所用的“高严谨性条件”包括等同于在42℃这样的溶液中结合和杂交的条件,所述溶液由5xsspe(43.8g/lnacl、6.9g/lnah2po4(h2o)和1.85g/ledta、用naoh将ph调节至7.4)、0.5%sds、5xdenhardt试剂和100mg/ml变性的鲑鱼精组成,然后当使用约500核苷酸长度的探针时,在42℃包括0.1xsspe、1.0%sds的溶液中洗涤。
在某些示例性实施方式中,鉴定细胞,然后分离单个细胞或多个细胞。本公开范围内的细胞包括任何类型的细胞,对于其中dna含量的理解被本领域技术人员认为是有用的。根据本公开的细胞包括任何类型的癌细胞、肝细胞、卵母细胞、胚胎、干细胞、ips细胞、es细胞、神经元、红细胞、黑素细胞、星形胶质细、生殖细胞、少突胶质细胞、肾细胞等。根据一个方面,本发明的方法使用来自单个细胞的细胞dna进行。多个细胞包括约2至约1,000,000个细胞,约2至约10个细胞,约2至约100个细胞,约2至约1,000个细胞,约2至约10,000个细胞,约2至约100,000个细胞,约2个至约10个细胞或约2至约5个细胞。
通过本文所述方法处理的核酸可以是dna,并且它们可以由任何有用的来源获得,例如人样品。在具体的实施方式中,双链dna分子被进一步定义为包含基因组,例如从来自人的样品获得的基因组。样品可以是来自人的任何样品,如血液、血清、血浆、脑脊液、脸颊刮擦物、乳头抽吸物、活组织检查、精液(可以称为射精液)、尿液、粪便、毛囊、唾液、汗液、免疫沉淀或物理分离的染色质等。在具体的实施方式中,样品包括单个细胞。在具体的实施方式中,样品仅包括单个细胞。
在特定实施方式中,由样品扩增和从头组装的核酸分子提供了诊断或预后信息。例如,由样品制备的核酸分子可提供基因组拷贝数和/或序列信息、等位基因变异信息、癌症诊断、产前诊断、亲子信息、疾病诊断、检测、监测和/或治疗信息、序列信息等。
本文所用“单个细胞”指一个细胞。可用于本文所述方法中的单个细胞可获自感兴趣组织,或活组织检查,血液样本,或细胞培养物。此外,可以获得来自特定器官、组织、肿瘤、赘生物等的细胞并将其用于本文所述的方法中。此外,通常,来自任何群体的细胞都可以用于所述方法中,如原核或真核单细胞生物体的群体,包括细菌或酵母。使用本领域已知的标准方法,可以获得单个细胞悬浮液,包括例如酶促使用胰蛋白酶或木瓜蛋白酶消化蛋白质,所述蛋白质在组织样品中连接细胞或在培养中释放贴壁细胞,或在样品中机械地分离细胞。可以将单细胞置于任何合适的反应容器中,在其中可以单独处理单个细胞。例如96孔板,从而将各单个细胞置于单个孔中。
用于操控单个细胞的方法是本领域已知的,并且包括荧光激活细胞分选术(facs)、流式细胞术(herzenberg.,pnasusa76:1453-551979)、显微操纵以及使用半自动细胞选择器(picker)(例如,来自stoelting有限公司的quixelltm细胞转移系统)。例如,可以基于通过显微镜观察可检测的特征(如位置、形态或报告基因表达)单独选择个体细胞。此外,还可以使用梯度离心和流式细胞术的组合来增加分离或分选效率。
一旦鉴定到所需细胞,使用本领域技术人员已知的方法将细胞裂解以释放包括dna的细胞内容物。细胞内容物被包含在容器或收集体积内。在本发明的一些方面,细胞内容物(如基因组dna)可通过裂解细胞从细胞释放。例如,裂解可以通过这样实现,加热细胞,或通过使用洗涤剂或其它化学方法,或通过这些的组合。然而,可以使用本领域已知的任何合适的裂解方法。例如,在存在吐温-20的情况下,于72℃加热细胞2分钟足以将细胞裂解。或者,可以将细胞于65℃水中加热10分钟(esumi等.,neuroscires60(4):439-51(2008));或于70℃在补充有0.5%np-40的pcr缓冲液ii(应用生物系统公司(appliedbiosystems))中90秒(kurimoto等.,nucleicacidsres34(5):e42(2006));获自裂解可以使用蛋白酶实现,如蛋白酶k,或通过使用离液盐,如异硫氰酸胍(美国公布号2007/0281313)。根据本文所述方法扩增基因组dna可以直接在细胞裂解物上进行,从而使得可以将反应混合物添加到细胞裂解物。或者,可以使用本领域技术人员已知的方法将细胞裂解物分成两个或更多个体积,如分到两个或更多个容器、管或区域,其中个体积容器、管或区域包含细胞裂解物的一部分。然后,通过本文所述方法或本领域技术人员已知的方法,可以扩增包含在各容器、管或区域中的基因组dna。
用于本发明的核酸还可以包括天然或非天然碱基。就此而言,天然脱氧核糖核酸可以具有选自腺嘌呤、胸腺嘧啶、胞嘧啶或鸟嘌呤的一个或多个碱基,并且核糖核酸可以具有选自尿嘧啶,腺嘌呤,胞嘧啶或鸟嘌呤的一个或多个碱基。可以包括在核酸中的示例性非天然碱基(不具有天然骨架或类似物结构)包括但不限于,肌苷,黄嘌呤(xathanine),次黄嘌呤(hypoxathanine),异胞嘧啶,异鸟嘌呤,5-甲基胞嘧啶,5-羟甲基胞嘧啶,2-氨基腺嘌呤,6-甲基腺嘌呤,6-甲基鸟嘌呤,2-丙基鸟嘌呤,2-丙基腺嘌呤,2-硫代尿嘧啶,2-硫代胸腺嘧啶,2-硫代胞嘧啶,15-卤代尿嘧啶,15-半胱氨酸,5-丙炔基尿嘧啶,5-丙炔基胞嘧啶,6-偶氮尿嘧啶,6-偶氮胞嘧啶,6-偶氮胸腺嘧啶,5-尿嘧啶,4-硫尿嘧啶,8-卤代腺嘌呤或鸟嘌呤,8-氨基腺嘌呤或鸟嘌呤,8-硫醇腺嘌呤或鸟嘌呤,8-硫代烷基腺嘌呤或鸟嘌呤,8-羟基腺嘌呤或鸟嘌呤,5-卤代尿嘧啶或胞嘧啶,7-甲基鸟嘌呤,7-甲基腺嘌呤,8-氮杂鸟嘌呤,8-氮杂腺嘌呤,7-脱氮鸟嘌呤,7-脱氮腺嘌呤,3-脱氮鸟嘌呤,3-脱氮腺嘌呤等。一个特定实施方式可以利用核酸中的异胞嘧啶和异鸟嘌呤以减少非特异性杂交,如美国专利号5,681,702中所述。
本文所用“引物”通常包括这样的天然或合成的寡核苷酸,其与多核苷酸模板形成双链体时能够用作核酸合成的起点(如测序引物)并从其3’末端沿模板延伸以形成延伸的双链体。在延伸过程中添加的核苷酸序列由模板多核苷酸的序列决定。通常,引物通过dna聚合酶延伸。引物通常具有这样范围内的长度:3-36个核苷酸、5-24个核苷酸或14-36个核苷酸。本发明范围内的引物还包括正交引物、扩增引物、构建引物等。成对的引物可以侧接于感兴趣的序列或一组感兴趣的序列。引物和探针可以按顺序简并或准简并。本发明范围内的引物结合相连的靶序列。“引物”可以被认为是短多核苷酸,通常具有游离的3'-oh基团,其通过与靶标杂交结合潜在地存在于感兴趣样品中的靶标或模板,并在此后促进与该靶标互补的多核苷酸的聚合。本发明的引物由核苷酸组成,其范围在17-30个核苷酸。在一方面,引物是至少17个核苷酸、又或者至少18个核苷酸、又或者至少19个核苷酸、又或者至少20个核苷酸、又或者至少21个核苷酸、又或者至少22个核苷酸、又或者至少23个核苷酸、又或者至少24个核苷酸、又或者至少25个核苷酸、又或者至少26个核苷酸、又或者至少27个核苷酸、又或者至少28个核苷酸、又或者至少29个核苷酸、又或者至少30个核苷酸、又或者至少50个核苷酸、又或者至少75个核苷酸又或者至少100个核苷酸。
“扩增”或“进行扩增”这样的表达指通过其将形成特定多核苷酸的额外或多个拷贝的过程。
使用本领域技术人员已知的方法,可以对根据本文所述方法扩增的dna进行测序和分析。使用本领域已知的多种测序方法可以确定感兴趣的核酸序列的序列,所述方法包括但不限于通过杂交测序(sbh),通过连接测序(sbl)(shendure等.(2005)science309:1728),定量增量荧光核苷酸加法测序(qifnas),逐步连接和切割,荧光共振能量转移(fret),分子信标,taqman报告探针消化,焦磷酸测序,荧光原位测序(fisseq),fisseq珠(美国专利号7,425,431),摇摆测序(pct/us05/27695),多重测序(美国序列号12/027,039,提交与2008年2月6日;porreca等(2007)nat.methods4:931),聚合菌落(polony)测序(美国专利号6,432,360、6,485,944和6,511,803,以及pct/us05/06425);纳米网格滚环测序(rolony)(美国系列号2008年5月4日提交)、等位基因特异性寡聚体连接试验(例如,寡聚体连接试验(ola),使用连接的线性探针和滚环扩增(rca)读出的单模板分子ola,连接的锁式探针,和/或使用连接的环状锁式探针和滚环扩增(rca)读出的单模板分子ola)等。也可以利用高通量测序方法,例如,使用诸如roche454、illuminasolexa、ab-solid、helicos、polonator平台等的平台。本领域已知各种基于光的测序技术(landegren等.(1998)genomeres.8:769-76;kwok(2000)pharmacogenomics1:95-100;以及shi(2001)clin.chem.47:164-172)。
扩增的dna可以通过任何合适的方法进行测序。具体而言,可以使用高通量筛选方法扩增的dna进行测序,如应用生物系统公司(appliedbiosystems)的solid测序技术或亿明达公司(illumina)的基因组分析仪.在本发明的一个方面,可以对扩增的dna进行鸟枪法测序。读数的数量可以是至少10,000、至少100万、至少1000万、至少1亿或至少10亿。在另一方面,读数的数量可以是10,000-100,000,或者100,000-100万,或者100万-1000万,或者1000万-1亿、或者1亿到10亿。“读数(read)”是通过测序反应获得的连续核酸序列的长度。
“鸟枪法测序”是指用于测序非常大量的dna(如整个基因组)的方法。在该方法中,首先将待测序的dna被切成较小的片段,可以对其进行单独测序。然后根据这些片段的重叠序列将这些片段的序列重组为它们的原始顺序,从而产生完整的序列。可以使用多种不同的技术来完成dna的“切碎”,包括限制酶消化或机械剪切。重叠序列通常由适当编程的计算机对齐。鸟枪法测序cdna文库的方法和程序在本领域中是公知的。
扩增和测序方法在预测医学领域是有用的,其中诊断试验、预后试验、药物基因组学和监测临床试验用于预后(预测)目的,从而预防性地治疗个体。相应地,本发明的一个方面涉及诊断试验,其用于确定基因组dna以便确定个体是否处于患病症和/或疾病的风险中。这样的试验可用于预后或预测目的,从而因此在病症和/或疾病发作之前预防性治疗个体。相应地,在某些示例性实施方式中,提供了使用本文所示一种或多种表达谱方法来诊断和/或预测一种或多种疾病和/或病症的方法。
本文所用术语“生物样品”旨在包括但不限于从对象中分离的组织、细胞、生物液体和分离物,以及对象中存在的组织、细胞和生物液体。
在某些示例性实施方式中,提供了包含本文所述的一种或多种基因组dna序列的电子设备可读介质。本文所用“电子设备可读介质”指用于存储、携带或保持可由电子设备直接读取和访问的数据或信息的任何合适的介质。这样的介质可以包括但不限于磁存储介质、如软盘,硬盘存储介质和磁带;光存储介质,如光盘;电子存储介质,如ram,rom,eprom,eeprom等;一般硬盘和这些类别的混合物,如磁/光存储介质。介质适用于或被配制成用于以使其上记录有本文描述的一个或多个表达谱。
本文所用术语“电子设备”旨在包括被配置成或适用于存储数据或信息的任何合适的计算或处理设备或其他设备。适合用于本发明的电子设备的示例包括独立计算设备;网络,包括局域网(lan)、广域网(wan)互联网、内联网和外联网;电子设备,如个人数字助理(pda)、蜂窝电话、寻呼机等;和本地和分布式处理系统。
本文所用“记录的”指用于在电子设备可读介质上存储或编码信息的处理。本领域技术人员可以容易地采用任何目前已知用于在已知介质上记录信息的方法来生成包含本文描述的一个或多个表达谱的制品。
可使用各种软件程序和格式来将本发明的基因组dna信息存储在电子设备可读介质上。例如,核酸序列可以用文字处理文本文件来表示,以如wordperfect和微软word等市售可得软件对其进行格式化,或以ascii文件的形式表示,存储在数据库应用程序,诸如db2、sybase、oracle等,以及以其他形式。可使用任何数量的数据处理器结构格式(例如,文本文件或数据库),从而获得或创建其上记录有本文所述一个或多个表达谱的介质。
应理解的是,已描述的本发明的实施方式仅用于说明本发明的一些应用和原理。基于本文的教导,本领域技术人员可进行多种修改而不偏离本发明的真正精神与范围。贯穿本发明中所引用的所有参考文献、专利和公开专利申请的内容通过引用全文纳入本文并用于所有目的。
以下实施例是本发明的代表。这些实施例并不构成对本发明范围的限制,因为这些和其他等价实施方式将对于本发明、附图和所附权利要求而言是显而易见的。
实施例i
一般方案
下述一般方案能够用于全基因组扩增。在裂解缓冲液中裂解单个细胞。将包含具有特异性条码对(如独特条码对)的转座体的转座体文库以及转座缓冲液添加到细胞裂解物,将其充分混合并在55℃孵育10分钟。转座后添加1mg/ml蛋白酶以从对单细胞基因组dna的接合去除转座酶。将deepvent外切dna(exo-dna)聚合酶、dntp、pcr反应缓冲液和引物添加到反应缓冲液,加热至72℃10分钟以填充转座子插入产生的缺口。将反应混合物上样至微流体装置以形成微滴。将包含单个细胞基因组dna模板、dna聚合酶、dntp、反应缓冲液和引物的液滴收集到pcr管。进行40-60个pcr反应循环以扩增单个细胞基因组dna。选择循环的数量以驱动液滴中的扩增反应至饱和。裂解液滴,并将扩增产物纯化用于进一步分析,如高通量深度测序。
实施例ii
制造具有转座子dna同源二聚体的转座体
为了产生具有转座子dna同源二聚体的转座体(即,在各转座子dna上具有相同条码序列的转座体),以及由此具有独特的相关条码的转座体文库,包含切割位点(例如,dna核酸酶切割位点)、引发位点、独特条码序列和转座酶结合位点的多个转座子dna与单个微粒或珠连接,因此单个微粒包含具有相同的独特条码序列并且没有其他条码序列的多个转座子dna。
如图6所示,图1示出的多个条码化的转座子dna经由接头连接微粒,如珠。还提供了切割部分或位点,因此转座子dna可以被切割或以其他方式从微粒去除。
如图7中示例性的阐述,产生微粒的文库,其中文库中的各微粒具有与其连接的具有其独特条码序列的多个转座子dna。考虑了数百万的微粒,其中各微粒具有其独特的相关条码序列。本文所述的方法提供了同时且不单独地制造数百万对称标引的转座体,即各转座体具有其自身独特的相关条码序列,因为转座体的各转座子dna是相同的,而在单个反应体积中产生的转座体的数量是以百万计的。制造条码化的转座体的方法述于wo2012/2061832,然而,这类材料和方法不同于本文所述的那些,并且导致可以制造的转座体数量有限。根据一个方面,文库的相同单个微粒上的所有转座子dna具有相同的条码序列,而文库中基本上各微粒或各微粒具有其独特的相关条码序列,即,各微粒包含具有条码序列的转座子dna,所述条码序列不同于文库中各其余的微粒。根据一个方面,特定微粒上转座子dna分子的数量超过将与转座子dna分子接触形成转座体的转座酶分子的数量。以此方式,各转座体将具有2个相同的转座体dna分子,并且因此也将在两个转座子dna分子的每一个中具有相同的条码序列。例如,具有比存在的转座酶分子更多的转座子dna分子保证在微滴内形成转座体的过程中,没有转座体将缺少转座子dna分子。因此,减少或清除具有2个不同转座子dna分子(和相应地,2个不同的条码序列)的转座体复合物的存在。
然后将珠上样到微滴以及转座酶和核酸酶,从而使各微滴仅包含一个珠,并因此仅包含一个独特的条码。在微滴内,由珠切割转座子dna,并且形成具有相同独特条码序列的转座体(即转座子dna同源二聚体)。然后在裂解或破裂液滴后收集具有同源二聚化转座子dna的转座体,以形成转座体文库。
具体地,为了制造超过1,000个各自携带其自身独特的相关条码序列的转座体,利用微粒或珠或液滴微流体。根据macosko等cell161(5),2015(通过引用其全部内容纳入本文)中所述的方法,合成m数量各自携带具有独特条码的dna链的微粒或珠,因此在共有相同条码的各微粒或珠上存在平均n数量的转座子dna链,所述相同条码与微粒或珠特异性地相关,并且各微粒或珠具有其自身的独特条码序列,所述独特条码序列不同于其他微粒或珠。每个转座子dna经由接头分子连接微粒或珠,并且其序列包含切割位点(例如,可以通过新英格兰实验室公司(newenglandbiolabs)的usertm酶切割的单个尿嘧啶核苷酸),引发位点,独特条码序列和转座酶结合位点,并且所有珠或微粒上的所有dna链共有切割位点的相同序列,引发位点的相同序列和转座酶结合位点的相同序列。然后将所有的微粒或珠与相同序列的单链dna分子混合,所述相同序列与珠或微粒上dna链上的转座酶结合位点互补,因此可以在珠或微粒上生成部分双链和部分单链的dna分子,如图6所述。因为转座体比单链dna更有效地插入双链dna,该部分单链dna结构可以防止转座体分子之间的插入。
为了制造独特的条码化转座体,使用流动聚焦微流体装置将各微粒或珠与转座酶和切割酶(如来自新英格兰实验室公司的usertm酶)共包封在液滴中,如macosko等cell,2015,161(5):1202-14页和klein等cell,2015,161(5):1187-1201页所述,各自通过引用将其全部内容纳入,从而使各液滴包含0到1个珠或微粒。示例性的流体回路如图8所述,其包括经由微通道流体连通的水相酶混合物入口,水相珠入口,疏水性液体入口(称之为油入口),用于合并珠与酶混合物的组合区,和用于合并水相与油相的组合区,其通过与乳液液滴出口区域的微通道进一步流体连通。酶混合物与珠合并,并且然后该组合形成微滴,其中每个微滴1个珠。
合适的疏水相是这样的,当水性介质导入疏水相时,其产生水性液滴。本领域技术人员已知合适的油相,其中水相自发地产生水性液滴或分离的体积或被油相围绕的隔室。示例性的疏水相包括疏水性液体,如油,如氟化油,如3-乙氧基全氟(2-甲基己烷),和表面活性剂。表面活性剂为本领域技术人员熟知。包含合适的油和表面活性剂的示例性疏水相可以下述购得:用于evagreen(伯乐公司(bio-rad))的qx200tm液滴生成油,包含疏水性表面活性剂的液体,其不与水性溶液混合或者在水性溶液中负面影响生化反应,hfe7500中的008-含氟表面活性剂(ran生物技术公司(ranbiotechnologies)),pico-surftm1(多洛麦特微流体公司(dolomitemicrofluidics)),专有油性表面活性剂(raindance技术公司(raindancetechnologies)),mazutis,l.,等,使用基于液滴的微流体的单细胞分析和分选(single-cellanalysisandsortingusingdroplet-basedmicrofluidics),natureprotocols,2013,8,870-891页中所讨论的氟化油中所述的含氟表面活性剂,和baret,j.-c.,labonachip,2012,12,422-433页中所述的其他表面活性剂,其各自通过引用其全部内容纳入本文。
当油相和水相在组合区或乳液液滴出口区域组合时,水相将自发地形成被油相包围的液滴。根据一个方面,疏水性流体如油的冲洗体积在微流体设计内或者用于将水性珠相或水性酶混合物相输入到微流体设计中的针筒或注射器内的水相上游用于输入可能以其他方式占据死体积的任何的水相,以使导入微流体芯片设计中的原始水相的损失最小化,疏水性流体如油可以不含冲洗体积不需要的表面活性剂。有用的微流体芯片设计可以使用autocad软件(欧特克有限公司(autodeskinc.))创建,并且可以通过cad工艺服务有限公司(cadartservicesinc.)打印成光掩膜用于微流体制造。可以使用mazutis等natureprotocols8(5),2013中所述的常规技术创建模具或母板,通过引用将其全部内容纳入本文。微流体芯片可由母板通过将未固化的聚二甲基硅氧烷(pdms)(道康宁sylgard184)浇注到母板上并加热至固化以形成具有沟槽或回路的表面来制造。形成入口和出口孔,并将具有回路的固化表面面对载玻片放置并固定以产生微通道和微流体芯片。使用之前,微流体芯片的内部可以用化合物处理,用于改善微流体芯片内部的疏水性并且洗涤去除可能的污染。
根据一个方面,使用本领域技术人员已知的常规方法生成液滴,其中各液滴包含单个珠或没有珠。将水性介质中的酶混合物和水性介质中的珠合并,并将组合物导入油中,产生液滴,其中液滴的数量超过珠的数量,从而在单个液滴内分离单个珠以及足够的酶。
各液滴内,通过切割酶从微粒或珠切割n数量的连接微粒的转座子dna分子,并在微粒内与转座酶单体同时组装成约n/2数量的转座体,其各自由具有相同条码的2个转座子dna分子和2个转座酶单体组成,如图7所示。将条码的数量(即包封的微粒或珠的数量(即m))以及液滴中转座体的平均数量(即各微粒或珠上dna链的平均数量的一半(即,n的一半))缩放,从而可以获得具有统计学上独特条码的转座体,用于在切割位点切割和插入或添加转座子dna,即邻接基因组dna片段的各末端。
具有有效的独特序列的转座体这样获得,通过汇集所有m乘以n/2数量的转座体,通过裂解液滴,即破乳作用,并收集转座体,并将转座体(具有m条码和平均n/2拷贝的各条码)集合总量的小部分(显著少于1/(n/2))插入基因组,从使具有含有相同条形码插入基因组的2个或更多个转座体的机会在统计学上是微小的。通过向液滴添加全氟辛醇(tci化学品公司(tcichemicals))并用手摇动或涡旋和离心后可以实现裂解液滴或破乳作用,所有的液滴被裂解并收集包含转座体的水性溶液。
作为非限制性示例,为了组装具有约6,000,000,000碱基对的人基因组,插入基因组需要1,000,000个具有独特条码的转座体,假设6000bp平均插入长度,那么m至少是106,例如其可以是107。考虑到典型的微粒或珠可以带有大约108个dna分子,如macosko等cell161(5),2015中解释,n=108是合理的估值。因此,如果使用m=107个微粒或珠制造5x1014(m乘以n/2)数量的条码化转座体,那么可以获取转座体全部集合的1/166667并添加到基因组dna,并且大约1/3000的添加的转座体可以插入基因组,因此插入基因组的转座体的最终数量预计为5x1014x1/166667x1/3000,其大约是1,000,000个。在该实施例中,插入基因组的转座体大约是1/500,000,000(1/166667乘以1/3000)这显著地小于1/(n/2),因此2个相同的条码插入基因组的机会在统计学上是微小的。简言之,为了使用6000bp的平均插入长度组装人基因组,1000万个独特条码化的珠可以用于制造条码化的转座体,并且在该实施例中,1/166667的总转座体需要被添加到基因组dna用于插入。
在一实施方式中,微粒或珠上各dna链的切割位点可以是这样的位点,其可以在uv光暴露后被切割,如klein,a.m.,等,用于应用于胚胎干细胞单细胞转录学的液滴条码化(dropletbarcodingforsingle-celltranscriptomicsappliedtoembryonicstemcells).cell,2015.161(5):1187-1201页中所述,通过引用其全部内容纳入本文。在该实施例中用于酶混合物的水相可能不包含用于从微粒或珠切割dna链的切割酶。
在另一实施方式中,条码化的珠(或颗粒或微粒)可以是多孔珠,以这样的方法dna分子可以结合在材料上或材料的多孔网络内。可以选择用于酶混合物的缓冲液,因此一旦珠与酶混合物共封装于液滴中后,结合在珠上或珠的孔内的dna可以从珠释放,并且随后与转座酶单体在液滴内组装成转座体。可以依赖于缓冲液条件的受控的方式携带并释放dna的材料和方法的示例包括gemcodetm颗粒(10xgenomics),包括核酸纯化试剂盒,如dnaclean&concentratortm-5(zymo研究公司(zymoresearch),monarch核酸纯化试剂盒(新英格兰生物实验室公司)和qiaquickpcr纯化试剂盒(凯杰公司(qiagen))中的离心柱,并且包括boom,r.等,用于核酸纯化的快速简单方法(rapidandsimplemethodforpurificationofnucleicacids).journalofclinicalmicrobiology,1990,28(3),495-503页;chen,c.w.和thomasjr.,c.a.从琼脂糖凝胶回收dna区段(recoveryofdnasegmentsfromagarosegels).analyticalbiochemistry,1980,101(2),339-341页;和tian,h.,等,评估二氧化硅树脂以小型化的形式从复杂的生物基质中直接高效地提取dna(evaluationofsilicaresinsfordirectandefficientextractionofdnafromcomplexbiologicalmatricesinaminiaturizedformat).analyticalbiochemistry,2000,283,175-191页中所述的材料和方法,其各自通过引用其全部内容纳入本文。
在一些方面,条码化的颗粒可以被条码化的液滴替换,所述条码化的液滴已经扩增并述于lan,f.,等,用于大量平行单分子深度测序的液滴条码化(dropletbarcodingformassivelyparallelsingle-moleculedeepsequencing)).naturecommunications,2016,7:11784,通过引用其全部内容将其纳入本文。然后使用中abate,a.,等,以微流体使用微注射的高通量注射(high-throughputinjectionwithmicrofluidicsusingpicoinjectors).proceedingsofthenationalacademyofsciencesoftheunitedstatesofamerica,2010,107(45),19163-19166页;lan,f.,等,用于大量平行单分子深度测序的液滴条码化(dropletbarcodingformassivelyparallelsingle-moleculedeepsequencing).naturecommunications,2016,7:11784;和rhee,m.,等,液滴微流体系统中用于可再现微注射的压力稳定器(pressurestabilizerforreproduciblepicoinjectionindropletmicrofluidicsystems).labonachip,2014,14(23),4533-4539页所述的微注射或液滴合并方法可以将酶混合物导入条码化的液滴,其各自通过引用将其全部内容纳入本文。然后,在各液滴内,导入的转座体单体可以与转座子dna分子组装成转座体,所述转座子dna分子具有液滴特异性条码。然后,可以裂解所有的液滴,从而可以汇集条码化的转座体,用于以条码化的注释插入基因组dna。
根据一个方面,本文所述具有转座子dna序列的转座体可以在并非使用液滴微流体产生的分开的隔室内合成;这类平台、设备、材料或方法的示例包括多孔板,高通量合成器,微阵列,微孔,微反应器或其他区室化方法,诸如sims,p.a.,等,pdms微反应器内荧光dna测序(fluorogenicdnasequencinginpdmsmicroreactors).naturemethods,2011,8(7),575-580页;gole,j.,等,使用纳升微孔的单个细胞的大量平行聚合酶克隆和基因组测序(massivelyparallelpolymerasecloningandgenomesequencingofsinglecellsusingnanolitermicrowells.naturebiotechnology,2013,31(12),1126-1132页;leungk.,等,平面底物上稳健的高通量纳升体积单细胞多项置换扩增(robusthigh-performancenanoliter-volumesingle-cellmultipledisplacementamplificationonplanarsubstrates).proceedingsofthenationalacademyofsciencesoftheunitedstatesofamerica.2016,113(30),8484-8489页;和zarzar,l.d.,等,通过可调节界面张力动态重新配置复合乳液(dynamicallyreconfigurablecomplexemulsionsviatunableinterfacetensions.nature,2015,518,520-524页中所述的那些,其各自通过引用其全部内容纳入本文。
实施例iii
细胞裂解
选择细胞,将其从培养皿上切下并使用激光解剖显微镜(lmd-6500,莱卡公司(leica))以如下方式分配到管中。将细胞铺板到膜包覆的培养皿上,并用10倍物镜明场显微镜(莱卡公司)观察。然后使用uv激光切割单独选择的细胞周围的膜,从而使其落入pcr管的帽中。对管进行短暂离心以使细胞下降至管的底部。向pcr管的侧面添加3-5μl的裂解缓冲液(30mmtris-clph7.8、2mmedta、20mmkcl、0.2%曲通x-100、500μg/mlqiagen蛋白酶)并向下离心。然后,在pcr仪器上使用如下温度方案对捕获的细胞进行热裂解:50℃3小时,75℃30分钟。或者,将单个细胞用口吸管吸移到含有edta和蛋白酶(如10-5000μg/ml浓度的qiagen蛋白酶(凯杰公司(qiagen)))的低盐裂解缓冲液中。孵育条件根据使用的蛋白酶变化。在qiagen蛋白酶的情况中,孵育是37-55℃下进行1–4小时。然后将蛋白酶加热至80℃失活,并进一步通过特异性蛋白酶抑制剂,如4-(2-氨基乙基)苯磺酰氟盐酸盐(aebsf)或苯基甲磺酰氟(pmsf)(西格玛奥德里奇公司(sigmaaldrich))使其失活。细胞裂解物保存在-80℃。
实施例iv
转座
单个细胞裂解物和转座体文库在含有1–100mmmg2+和任选地含有1–100mmmn2+或co2+或ca2+的缓冲液系统中混合,并于37-55℃孵育5-240分钟。反应体积根据细胞裂解物体积变化。反应中添加的转座体文库的量可根据所需片段化尺寸随时调整。通过使用edta和任选地egta或其它离子螯合剂来螯合mg2+以停止转座反应。任选地,可以向混合物添加短双链dna作为加标(spike-in)。残留转座体通过蛋白酶消化失活,如以1-500μg/ml最终浓度的qiagen蛋白酶在37-55℃下进行10-60分钟的蛋白酶消化。然后通过加热和/或蛋白酶抑制剂如aebsf使蛋白酶失活。
实施例v
缺口填平
转座和转座酶去除后,将包含mg2+、dntp混合物、引物和热稳定dna聚合酶如deepvent外切-dna聚合酶(新英格兰生物实验室公司)的pcr反应混合物在合适的温度下添加到溶液,并持续合适的时间段以通过填充转座反应留下的9bp的缺口。缺口填平孵育温度和时间取决于所用特异性dna聚合酶。反应后,通过加热和/或蛋白酶处理,如qiagen蛋白酶,使dna聚合酶任选地失活。如果使用蛋白酶,其后通过加热和/或蛋白酶抑制剂使其失活。
实施例vi
dna片段扩增
根据一个方面,使用本领域技术人员已知的常规方法扩增dna片段。将来自上述实施例的缺口填平的双链产物,包括具有引物结合位点的dna片段添加到水性介质中的pcr反应试剂。然后,水性介质按照pcr条件对各dna片段进行pcr扩增。
实施例vii
使用条码对dna片段扩增子进行测序和基因组从头组装
根据一个方面,使用本领域技术人员已知的方法对片段进行测序,并将序列储存在计算机可读储存器中。然后可以比较序列,并且可以鉴定具有匹配条码序列的片段。然后将具有匹配条码序列的片段鉴定为在原始基因组dna序列中彼此相邻的序列。然后可以将2个或更多个邻接序列计算连接在一起,即在计算机中使用计算机软件,以产生原始基因组dna的较长序列片段。在该方面中,本公开提供了从头组装使用转座体条码产生的基因组dna片段以产生较长片段的方法。
根据一个方面,每个基因组dna片段的各末端具有缺口填平的序列,以及转座酶结合位点序列,条码序列和引发序列。缺口填平的序列可以作为用于将不同的片段栓链到较长基因组序列中的第二组条码,因为其是被转座体切割的2个片段的重复序列。例如,已知的是当tn5转座体插入双链基因组dna模板时,其在插入位点的两个末端各自留下单链9bp的缺口,如图3所示,并且在图4所示的缺口填平步骤(也称之为缺口延伸步骤)后,相同插入位点上两个9bp的缺口将共有相同的序列。插入位点上这类重复的9bp的序列可以作为额外的条码,用于栓链用于从头组装的片段,其在当插入携带相同条码序列的2个转座体发生时特别有用。
根据一个方面,片段在计算机中通过匹配条码序列从头组装以再生原始基因组dna序列,如全基因组dna。在使用条码信息将所有片段栓链后,由片段组成的栓链的、连接的或组装的连续或领接的基因组序列(也称之为“毗连群”)可以与另一毗连群比较或匹配,所述另一毗连群与同源染色体共有相似的或相同的序列,并且通过匹配来自同源染色体的毗邻群,基因组序列或毗邻群可以进一步连接成较长的序列或毗邻群,最终将其组装成完整的基因组。本领域技术人员已知的从头组合方法包括重叠-布局-共有(overlap-layout-consensus)(olc),debruijn,字符串图表方法和chaisson,m.j.p.等,遗传变异和人类基因组的从头组装(geneticvariationandthedenovoassemblyofhumangenomes).naturereviewgenetics,2015.16:627-640页中综述的其他组装算法,其通过引用将其全部内容纳入本文用于所有目的。
根据一个方面,可以条码化的注释独立地片段化和扩增来自2、3、4或更多个子细胞或相同细胞的基因组,测序,使用前述的方法分开地组装以有效地提供基本上同源的染色体对用于交叉引用,从而获得独特的从头组装的基因组图谱。这些方法可以与这样的从头组装方法合并,所述从头组装方法利用同源物,诸如miller等genomics,95(6),2010中综述的ssake、sharcgs、vcake、newbler、celeraassembler、euler、velvet、abyss、allpaths和soapdenovo之间的重叠区域;以及chaisson等naturereviewsgenetics,16,2015中所述的算法,上述文献各自通过引用将其全部内容纳入本文,以提供基本上同源的重叠,用于高质量全基因组从头组装。
当靶基因组dna来自具有超过一个倍性的单个细胞时,基因组的从头组装还可以实现单体型分析,如图9所示。倍性是细胞中染色体组的数量。例如,人体细胞具有各染色体的2组同源拷贝。这2个拷贝或等位基因分别来自父亲和母亲,并且在细胞中是2个物理上隔离的dna。因为这2个拷贝没有连接在一起,并且使用转座体进行转座,即插入转座子dna和产生片段对于每个单独的拷贝独立地发生,一个拷贝的任何部分不与另一拷贝的任何部分共有相同的插入位点,所以来自一个拷贝的片段不包含可以匹配另一拷贝的片段上任何条码的条码,并且因此来自一个拷贝的片段将不会连接或栓链于来自另一拷贝的那些片段。例如,如图9所示,转座体1和2将转座子dna插入第一等位基因,而转座体3和4将转座子dna插入第二等位基因。在独立转座体启动对于每个单独的等位基因插入转座子dna后以及使用本文所述方法扩增、测序和从头组装后,单独地组装2个等位基因,并且最终组装的产物是单体型解析(resolved)的基因组。这是因为等位基因1的片段不具有与来自等位基因2的任何片段相同的条码。所以来自等位基因1的片段不会连接或栓链来自等位基因2的那些片段,并且各等位基因内的片段可以独立于来自等位基因2的任何信息连接或栓链,反之亦然。相应地,产生的从头组装将在栓链的序列中产生更长的链,并且来自相同等位基因的全染色体组装,并且因此,基因组dna是单体型解析的。相反,当人基因组通过鸟枪测序组装时,它被视为单倍体基因组,因为2个等位基因几乎相同且无法区分。使用本文所述的转座体方法,因为与各等位基因相关的独特条码序列,如9所示,单独地组装2组染色体。该方法允许通过匹配条码逐一连接所有等位基因1片段,以及通过匹配条码逐一连接所有等位基因2片段,区分等位基因2与等位基因1。独特条码的组装导致从头组装单独的等位基因,其产生单体型解析。
实施例xi
试剂盒
公开的扩增方法所需的材料和试剂可在试剂盒中组装在一起。本公开的试剂盒通常将至少包括进行所要求保护的方法所需的转座体(由转座酶和转座子dna组成)、核苷酸和dna聚合酶、与所需引物组。在优选实施方式中,试剂盒还将包括用于由dna样品扩增dna的说明。示例性的试剂盒是那些适合用于扩增全基因组dna的试剂盒。在各种情况中,试剂盒将优选具有对各种单独试剂、酶或反应物不同的容器。通常,将各物质在其各自的容器中适当分装。试剂盒的容器装置通常包括至少一个小瓶或试管。也可以是能够将试剂放置并分装其中的细颈瓶、瓶子和其他容器装置。试剂盒的单个容器将优选保持密闭状态以用于商业销售。合适的较大容器可包括注塑或吹塑的塑料容器,其中保留所需小管。优选说明书与试剂盒一起提供。
实施例xii
实施方式
本公开提供了制造转座体文库的方法,其包括下述步骤:将多个转座子dna与多个微粒中的各微粒连接,其中连接单个微粒的所有转座子dna包括与所述单个微粒相关的共同独特条码序列,从而使所述多个微粒中的各微粒具有独特的相关条码序列,将具有与之连接的转座子dna的所述多个微粒与转座酶和切割酶合并以形成水性混合物,将所述水性混合物与油相混合,从而形成多个微滴,其中所述多个微滴中的各微滴在相应的单个微滴中与所述转座酶和所述切割酶一起分离,对于各相应的单个微滴,由所述相应的单个微滴内的微粒切割所述多个转座子dna,并在所述微滴内形成数个转座体,并且所述微滴内的各转座体具有含有所述共同独特条码序列的2个转座子dna,裂解所述多个微滴中的各微滴,和收集所述转座体以产生所述转座体文库。根据一个方面,转座体文库包括大于1,000个转座体。根据一个方面,转座体文库包括大于10,000个转座体。根据一个方面,转座体文库包括大于100,000个转座体。根据一个方面,转座体文库包括大于1,000,000个转座体。根据一个方面,转座体文库包括大于2,000,000个转座体。根据一个方面,转座体文库包括大于3,000,000个转座体。根据一个方面,转座体文库包括大于4,000,000个转座体。根据一个方面,转座体文库包括大于5,000,000个转座体。根据一个方面,转座体文库包括大于10,000,000个转座体。根据一个方面,该方法还包括提取一部分所述转座体文库以形成试剂转座体文库,其中所述试剂转座体文库的各转座体具有独特的相关条码序列。根据一个方面,该方法还包括提取一部分所述转座体文库以形成试剂转座体文库,其中所述试剂转座体文库内基本上所有的转座体具有独特的相关条码序列。根据一个方面,各转座子dna包含特异性引物结合位点和双链转座酶结合位点。根据一个方面,所述转座子dna包含双链转座酶结合位点和突出端,其中所述突出端包含位于所述突出端5'末端的引物结合位点和条码序列。根据一个方面,各转座子dna通过接头和切割位点与相应的微粒连接。根据一个方面,各转座子dna包括5'突出端并且在其相应的5'末端通过接头和切割位点与相应的微粒连接。根据一个方面,所述转座酶是tn5转座酶、mu转座酶、tn7转座酶或is5转座酶。根据一个方面,油相包含表面活性剂。根据一个方面,所述油相内的所述多个微滴通过将所述水性混合物与所述油相以产生比存在的微粒更多的微滴的方式合并产生。根据一个方面,所述油相内的所述多个微滴通过将所述水性混合物与所述油相以产生比存在的微粒更多的微滴的方式合并产生,并且其中所述多个微滴同时产生。根据一个方面,所述油相内的所述多个微滴通过将所述油相和所述水性介质在微流体芯片内合并产生。根据一个方面,多个微滴被破乳剂裂解。
本公开提供了基因组dna从头组装的方法,其包括步骤:将基因组dna与转座体的文库接触,所述文库的各转座体具有其独特的相关条码序列,其中所述文库的各转座体包含转座酶和转座子dna同源二聚体,其中所述同源二聚体的各转座子dna包含转座酶结合位点、独特条码序列和引物结合位点,其中所述转座体的文库结合沿着所述基因组dna的靶位置,并且所述转座酶将所述基因组dna切割成代表基因组dna片段文库的多个双链基因组dna片段,其中各双链基因组dna片段在所述基因组dna片段的各末端包含独特条码序列对的成员之一,对所述转座子dna和所述基因组dna片段之间的缺口进行缺口填平,以形成各末端具有引物结合位点的双链基因组dna片段延伸产物的文库,扩增所述双链基因组dna片段延伸产物以产生扩增子,对所述扩增子进行测序,和通过条码匹配将所述扩增子计算连接在一起,从而从头组装基因组dna。根据一个方面,基因组dna是获自单个细胞的全基因组dna。根据一个方面,所述转座酶是tn5转座酶、mu转座酶、tn7转座酶或is5转座酶。根据一个方面,所述转座子dna包含双链19bptnp结合位点和突出端,其中所述突出端包含位于所述突出端5'末端的引物结合位点和条码序列。根据一个方面,在缺口填平和延伸所述双链基因组dna片段之前,将结合的转座酶从所述双链片段去除。根据一个方面,所述转座酶是各自与转座子dna复合的tn5转座酶,其中所述转座子dna包含双链19bptnp结合位点和突出端,其中所述突出端包含条码序列和引物结合位点。根据一个方面,基因组dna来自产前细胞。根据一个方面,基因组dna来自癌细胞。根据一个方面,基因组dna来自循环肿瘤细胞。根据一个方面,基因组dna来自单个产前细胞。根据一个方面,基因组dna来自单个癌细胞。根据一个方面,基因组dna来自单个循环肿瘤细胞。根据一个方面,所述引物结合位点是特异性pcr引物结合位点。根据一个方面,所述从头组装是单体型解析的从头组装。根据一个方面,所述单体型解析的从头组装是在人白细胞抗原区,v(d)j重组区或人单个细胞的其他区域上。
本公开提供了基因组dna从头组装的方法,其包括步骤:产生含于非水相内的多个水性微滴,其中各微滴包含在所述微滴内形成的多个转座体,并且所有的转座体具有2个转座酶和2个相同的转座子dna,并且各转座子dna具有转座酶结合位点、条码序列和引物结合位点,释放各微滴中的所述多个转座体,并将所述释放的转座体收集到转座体文库中,在反应体积内形成试剂转座体文库,其中所述试剂转座体文库内基本上所有或所有转座体具有独特的相关条码序列,将基因组dna与试剂转座体文库在所述反应体积内接触,其中所述转座体结合沿着所述基因组dna的靶位置并且所述转座酶将所述基因组dna切割成代表基因组dna片段文库的多个双链基因组dna片段,并且各双链基因组dna片段包含所述基因组dna片段各末端上独特条码序列对的成员之一,对所述转座子dna和所述基因组dna片段之间的缺口进行缺口填平,以在所述反应体积内形成各末端具有引物结合位点的双链基因组dna片段延伸产物的文库,扩增所述双链基因组dna片段延伸产物以在所述反应体积内产生扩增子,对所述反应体积内的所述扩增子进行测序,和通过条码匹配将所述扩增子计算连接在一起,从而从头组装基因组dna。根据一个方面,试剂转座体文库包括大于1,000个转座体。根据一个方面,试剂转座体文库包括大于10,000个转座体。根据一个方面,试剂转座体文库包括大于100,000个转座体。根据一个方面,试剂转座体文库包括大于1,000,000个转座体。根据一个方面,试剂转座体文库包括大于2,000,000个转座体。根据一个方面,试剂转座体文库包括大于3,000,000个转座体。根据一个方面,试剂转座体文库包括大于4,000,000个转座体。根据一个方面,试剂转座体文库包括大于5,000,000个转座体。根据一个方面,试剂转座体文库包括大于10,000,000个转座体。根据一个方面,基因组dna是获自单个细胞的全基因组dna。根据一个方面,所述转座酶是tn5转座酶、mu转座酶、tn7转座酶或is5转座酶。根据一个方面,所述转座子dna包含双链19bptnp结合位点和突出端,其中所述突出端包含位于所述突出端5'末端的引物结合位点和条码序列。根据一个方面,在缺口填平和延伸所述双链基因组dna片段之前,将结合的转座酶从所述双链片段去除。根据一个方面,所述转座酶是各自与转座子dna复合的tn5转座酶,其中所述转座子dna包含双链19bptnp结合位点和突出端,其中所述突出端包含条码序列和引物结合位点。根据一个方面,基因组dna来自产前细胞。根据一个方面,基因组dna来自癌细胞。根据一个方面,基因组dna来自循环肿瘤细胞。根据一个方面,基因组dna来自单个产前细胞。根据一个方面,基因组dna来自单个癌细胞。根据一个方面,基因组dna来自单个循环肿瘤细胞。根据一个方面,所述引物结合位点是特异性pcr引物结合位点。
本公开提供了基因组dna从头组装的方法,其包括步骤:将转座酶与多个转座子dna在物理隔离的反应腔室内接触以在各物理隔离的反应腔室内形成转座体,其中各转座子dna包含共同转座酶结合位点、共同引物结合位点和条码序列,其中同一反应腔室内所有转座子dna的条码序列是相同的,但是与其他反应腔室内的转座子dna不同,收集各反应腔室的所述转座体,并混合所有所述转座体以形成转座体文库,在反应体积内形成试剂转座体文库,其中所述试剂转座体文库内基本上所有或所有转座体具有独特的相关条码序列,将基因组dna与试剂染色体文库在所述反应体积内接触,其中所述转座体结合沿着所述基因组dna的靶位置并且所述转座酶将所述基因组dna切割成代表基因组dna片段文库的多个双链基因组dna片段,并且各双链基因组dna片段包含所述基因组dna片段各末端上独特条码序列对的成员之一,对所述转座子dna和所述基因组dna片段之间的缺口进行缺口填平,以在所述反应体积内形成各末端具有引物结合位点的双链基因组dna片段延伸产物的文库,扩增所述双链基因组dna片段延伸产物以在所述反应体积内产生扩增子,对所述反应体积内的所述扩增子进行测序,和通过条码匹配将所述扩增子计算连接在一起,从而从头组装基因组dna。根据一个方面,所述反应腔室是管,多孔板,微阵列芯片,微孔,微反应器,微滴,微粒水凝胶或其他区室化方法。
- 还没有人留言评论。精彩留言会获得点赞!