一种新型甲基化建库方法及其应用与流程
本发明涉及分子生物学
技术领域:
,特别是涉及一种新型甲基化建库方法及其应用。
背景技术:
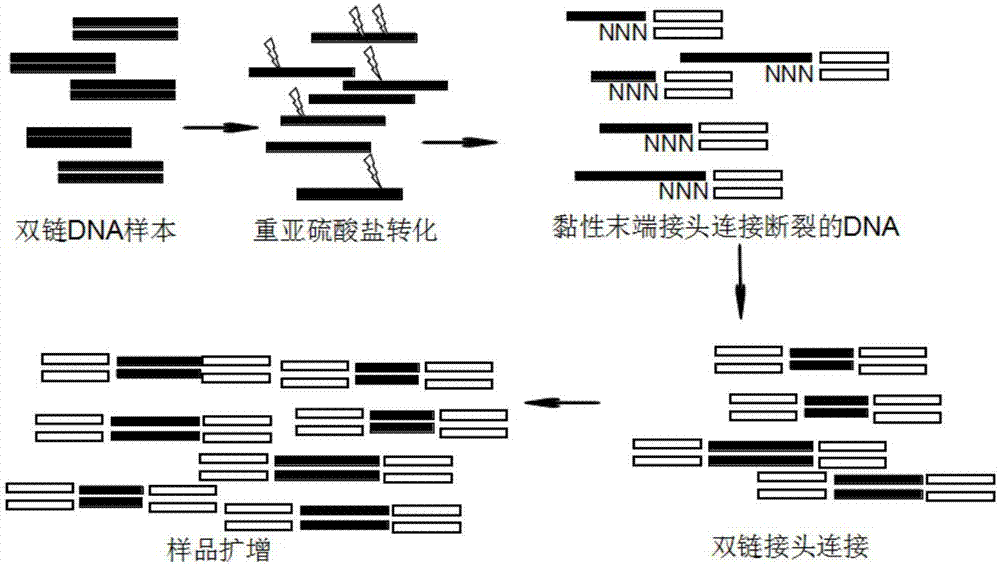
:甲基化是最常见的表观遗传修饰方式之一,在不改变核酸序列的前提下,调控基因的表达。DNA甲基化通常是指在DNA甲基转移酶的介导下,在胞嘧啶的第5位碳原子上加上一个甲基基团,使之变成5-甲基胞嘧啶(5-mC)。甲基化修饰在基因印迹、基因表达调控、X染色体失活、癌症发生与发展等方面发挥重要作用。DNA甲基化异常可通过影响原癌基因和抑癌基因的表达而参与肿瘤的形成。当肿瘤发生时,抑癌基因通常发生整体的高甲基化,尤其是CpG岛区域,从而抑制基因的表达,从而导致癌症的发生。另一方面原癌基因则发生大规模的去甲基化,导致原癌基因的激活而导致基因表达量的升高,进一步是癌症发生或恶化。近年来测序技术发展迅猛,已成为生物学研究的重要手段。高通量测序技术的兴起,使大规模、低成本研究DNA序列以及DNA序列的表观修饰成为可能。以甲基化修饰为代表的DNA的表观修饰一直以来都是研究热点。目前对于甲基化的研究方式们通常还是通过双链连接接头后进行重亚硫酸盐转化后扩增的方式进行。该方案对样品的起始量有着较高的要求,对于组织样品通常需要微克(μg)级的DNA起始量。但是由于甲基化的研究中的金标准--重亚硫酸盐处理,需要将DNA变性成单链,并且会造成样品的随机断裂,而造成文库信息的极大损失。专利“甲基化DNA检测方法”(公开号:106755319A),提出采用单链连接或环化的方式进行cfDNA样品的甲基化研究,该方案能对经过重亚硫酸转化的DNA进行较好的扩增,但是该方案中所需的酶(circligase)价格昂贵,若用其他酶进行替代则会造成连接效率的极大下降。另外该方案的滚环扩增或反向PCR扩增后,若需进行文库构建,则需要进行常规的文库构建流程,操作繁琐。SwiftBiosciences公司的SWIFTAccel-NGS-Methyl-Seq方法,提供的也是单链连接的方式进行文库构建。如专利“甲基化DNA检测方法”(公开号:106755319A)中所述,该方案的链接效率低,并且该商业化试剂盒的价格昂贵。目前对于甲基化样品进行扩增的实验方案主要包括下表中列举的双链接头连接扩增、单链环化、单链接头连接、随机引物扩增等,具体方案见表一。表一常用的DNA甲基化扩增及建库实验方案以上现有技术的缺点如下:一、双链接头连接双链接头连接的方式,需要的DNA长需要达到微克(μg)级,这对许多珍稀样品,是无法实现的。另外双链连接的方式需要在加接头后进行重亚硫酸盐的处理,该处理需要DNA为单链模式方能发挥作用,同时会对DNA造成随机的断裂损伤。这部分断裂的DNA在后续的PCR扩增中是无法进行扩增的,这就造成了信息不可逆的丢失。二、单链接头连接单链接头连接的方式进行甲基化文库的构建存在连接效率低,起始量高等问题。三、随机引物扩增法由于随机引物在结合到目的片段的时候具有一定的片段选择性,因此会造成最终样品的偏向性。另外随机引物的扩增方案由于引物结合的随机性,造成了再比对时只能采用单端比对的模式比对到参考基因组,导致比对精确性的下降。四、自身连接成环的扩增方法自身连接成环的方式需要使用circligase,第一,该酶会优先进行分子内的环化,因此如果需要接入接头进行预扩增的话,则相对链接效率会有极大的下降。第二,该酶对于目标分子的连接是有序列选择性的,对于3’末端是C的分子没有连接能力,对于5’末端是A\T\C的分子的连接效率要远远小于5’末端是G的分子,因此存在严重的偏向性。第三,该酶价格昂贵。针对以上问题,急需一种新型甲基化建库方法及产品。技术实现要素:鉴于以上所述现有技术的缺点,本发明的目的在于提供一种新型甲基化建库方法及其应用。为实现上述目的及其他相关目的,本发明的第一方面提供了一种甲基化建库方法,包括如下步骤:(1)对样本的基因组DNA进行重亚硫酸盐转化;(2)对步骤(1)中重亚硫酸盐转化后的样本基因组DNA进行处理,使其维持单链DNA状态;(3)将步骤(2)中获得的单链DNA与黏性末端接头进行连接,获得一端带有接头的单链DNA;(4)对步骤(3)中获得的一端带有接头的单链DNA进行延伸,形成一端带有接头的双链DNA;(5)采用双链接头连接到步骤(4)所得一端带有接头的双链DNA的另一端,形成双端带有接头的双链DNA;(6)对步骤(5)获得的双端带有接头的双链DNA进行扩增,获得甲基化测序文库。进一步地,所述步骤(1)中,所述样本基因组DNA来自单细胞或多细胞。进一步地,所述步骤(1)中,所述样本基因组DNA由单细胞样本裂解获得或由多细胞样本抽提获得。此外,可以是在进行步骤(1)之前,先对单细胞样本裂解或对多细胞样本抽提,从而获得所述样本基因组DNA。也可以采用由他人利用本领域所熟知的技术已经由单细胞样本裂解获得或由多细胞样本抽提获得的样本基因组DNA。进一步地,当样本的细胞数目在十万个以下时,可直接裂解获得样本基因组DNA,而无需进行抽提。所述细胞可以是原核细胞或真核细胞。所述真核细胞可以是植物细胞或动物细胞及微生物。所述动物细胞具体选自组织消化的细胞、培养所得的细胞、胚胎发育早期的细胞、癌症早期的细胞、未经富集培养的微生物细胞、微流控分选获得的细胞、流式分选获得的细胞、有限稀释获得的细胞、激光捕获等方法获得的细胞中的任一种。进一步地,所述样本基因组DNA的质量可以大于等于6pg。进一步地,所述步骤(1)中,本领域技术人员可采用公知的重亚硫酸盐转化试剂对样本基因组DNA进行重亚硫酸盐转化。进一步地,所述步骤(2)中,本领域技术人员可采用公知的技术,例如高温变性后猝冷、NaOH溶液碱变性等方案使重亚硫酸盐转化后的样本基因组DNA维持单链DNA状态。所述高温一般是指60℃-98℃。优选地为80℃-98℃。更优选地为90℃-98℃。处理时间一般为30s-10min。所述NaOH溶液为0.2~0.6MNaOH,1~1.5MNaCl。优选地为0.3~0.5MNaOH,1~1.5MNaCl。更优选地为0.4MNaOH,1.5MNaCl。进一步地,所述步骤(3)中,所述黏性末端接头为带有黏性末端的双链接头,所述黏性末端接头的一端为第一黏性末端,所述第一黏性末端至少包括随机序列。进一步地,所述随机序列的长度为2-30nt,优选的为4-20nt,更优选的为5-10nt。需要说明的是,所述随机序列用于与样本基因组DNA变性成的单链DNA通过互补配对进行连接。所述黏性末端接头中的双链部分的序列可以适用于相应的测序平台,如illumina测序平台。进一步地,所述黏性末端接头的另一端可以是平末端,也可以是第二黏性末端。当所述黏性末端接头的另一端为第二黏性末端时,所述第二黏性末端为任意序列或A/T/C/G寡核苷酸的重复序列,优选的为重复序列,长度优选为2-10nt,更优选为2-5nt。进一步地,所述第一黏性末端包括随机序列及特殊序列。进一步地,所述特殊序列为序列确定的序列。进一步地,所述第一黏性末端的长度为2-30nt,优选的为4-20nt,更优选的为5-10nt。进一步地,所述黏性末端接头中,带有第一黏性末端的单链的长度为10-70nt。优选为10-30nt。再优选为10-25nt。进一步地,所述黏性末端接头中,带有第一黏性末端的单链的退火温度为10-100℃。优选为20-80℃。再优选为30-60℃。进一步地,所述黏性末端接头中,带有第一黏性末端的单链的5’端进行磷酸化修饰。进一步地,所述黏性末端接头中,带有第一黏性末端的单链的5’端进行间臂(spacer)修饰。优选地,所述带有第一黏性末端的单链的5’端可采用连续的SpacerC12或C18Spacer或C3Spacer-CPG或C3Spacer或C9Spacer的修饰,Spacer可随意组合以增长碳链长度。进一步地,所述带有第一黏性末端的单链的5’端进行氨基修饰。如DMS(O)MT-Amino-Modifier-C6、Amino-Modifier-C3-TFA、Amino-Modifier-C12、Amino-Modifier-C6-TFA(trifluoroaceticacidprotectinggrouponamine)、Amino-dT、Amino-Modifier-5、Amino-Modifier-C2-dT、Amino-Modifier-C6-dT,氨基修饰可以随意组合。进一步地,所述带有第一黏性末端的单链的5’端既进行间臂(spacer)修饰又进行氨基修饰,间臂(spacer)修饰和氨基修饰可以随意组合。进一步地,所述带有第一黏性末端的单链的5’端采用甲基化修饰的重复A/T/C/G序列与C3Spacer或C6Spacer或Spacer12或Spacer12或SpacerC18的修饰的组合。优选地,采用甲基化修饰的重复A/T/C/G序列个数为2-8个为佳。进一步地,所述带有第一黏性末端的单链的3’端进行生物素化(bioten)修饰。进一步地,所述带有第一黏性末端的单链的3’端进行间臂(spacer)修饰。优选地,所述带有第一黏性末端的单链的5’端可采用连续的SpacerC12或C18Spacer或C3Spacer-CPG或C3Spacer或C9Spacer的修饰,Spacer可随意组合以增长碳链长度。进一步地,所述带有第一黏性末端的单链的3’端进行氨基修饰。如DMS(O)MT-Amino-Modifier-C6、Amino-Modifier-C3-TFA、Amino-Modifier-C12、Amino-Modifier-C6-TFA(trifluoroaceticacidprotectinggrouponamine)、Amino-dT、Amino-Modifier-5、Amino-Modifier-C2-dT、Amino-Modifier-C6-dT,氨基修饰可以随意组合。进一步地,所述带有第一黏性末端的单链的3’端既进行间臂(spacer)又进行氨基修饰,间臂(spacer)和氨基修饰可以随意组合。进一步地,所述带有第一黏性末端的单链的3’端既有间臂(spacer)修饰又进行氨基修饰和生物素化(bioten)修饰。优选地,所述间臂(spacer)修饰和氨基修饰位于所述单链的3’末端序列与生物素化(bioten)修饰之间。再优选地,所述间臂(spacer)修饰和氨基修饰与生物素之间用TEG进行连接。进一步地,所述带有第一黏性末端的单链的3’端的随机序列之后还可以采用AmC3或AmC6或AmC7的氨基修饰或spacer修饰。进一步地,所述带有第一黏性末端的单链还可以采用AmC3或AmC6或AmC7的修饰或硫代修饰。进一步地,所述黏性末端接头中,不带有第一黏性末端的那条单链称为另一条单链。进一步地,所述黏性末端接头中,另一条单链的长度为10-70nt。优选为10-30nt。再优选为10-25nt。进一步地,所述黏性末端接头中,另一条单链的退火温度为10-100℃。优选为20-80℃。再优选为30-60℃。进一步地,所述黏性末端接头中,另一条单链的5’端进行磷酸化修饰。进一步地,所述黏性末端接头中,另一条单链的5’端进行间臂(spacer)修饰。优选地,采用C3Spacer或C6Spacer或SpacerC12或Spacer12或SpacerC18或C3Spacer-CPG的修饰,Spacer可随意组合以增长碳链长度。再优选地,所述黏性末端接头中,另一条单链的5’端采用1-10个SpacerC12或1-10个C18Spacer或1-10个C3Spacer或1-10个C6Spacer或1-10个C3Spacer-CPG的修饰。再优选地,所述黏性末端接头中,另一条单链的5’端采用甲基化修饰的重复A/T/C/G序列与C3Spacer或C6Spacer或Spacer12或Spacer12或SpacerC18的修饰的组合。优选地,采用甲基化修饰的重复A/T/C/G序列个数为2-8个为佳。进一步地,所述黏性末端接头中,另一条单链的5’端进行氨基修饰。如DMS(O)MT-Amino-Modifier-C6、Amino-Modifier-C3-TFA、Amino-Modifier-C12、Amino-Modifier-C6-TFA(trifluoroaceticacidprotectinggrouponamine)、Amino-dT、Amino-Modifier-5、Amino-Modifier-C2-dT、Amino-Modifier-C6-dT。进一步地,所述黏性末端接头中,另一条单链的3’端进行生物素化(bioten)修饰。进一步地,所述黏性末端接头中,另一条单链的3’端进行间臂(spacer)修饰。优选地,所述黏性末端接头中,另一条单链的5’端可采用连续的SpacerC12或C18Spacer或C3Spacer-CPG或C3Spacer或C9Spacer的修饰,Spacer可随意组合以增长碳链长度。进一步地,所述黏性末端接头中,另一条单链的的3’端进行氨基修饰。如DMS(O)MT-Amino-Modifier-C6、Amino-Modifier-C3-TFA、Amino-Modifier-C12、Amino-Modifier-C6-TFA(trifluoroaceticacidprotectinggrouponamine)、Amino-dT、Amino-Modifier-5、Amino-Modifier-C2-dT、Amino-Modifier-C6-dT,氨基修饰可以随意组合。进一步地,所述黏性末端接头中,另一条单链的3’端既进行间臂(spacer)修饰又进行氨基修饰,间臂(spacer)修饰和氨基修饰可以随意组合。进一步地,所述黏性末端接头中,另一条单链的3’端既有间臂(spacer)修饰又进行氨基修饰又有生物素化(bioten)修饰。优选地,所述间臂(spacer)修饰和氨基修饰位于所述第二引物序列的3’末端端序列与生物素化(bioten)修饰之间。再优选地,所述间臂(spacer)和氨基修饰与生物素之间用TEG进行连接。进一步地,所述步骤(3)中,所述黏性末端接头通过连接酶连接到所述单链DNA上。进一步地,所述连接酶选自HiFiTaqDNA连接酶、T4RNA连接酶2、连接酶、9°NTMDNA连接酶、TaqDNA连接酶、T7DNA连接酶、T3DNA连接酶、电转连接酶、平末端/TA连接酶预混液、瞬时黏性连接酶预混液、T4DNA连接酶、T4RNA连接酶、CircligasessDNAligase、5′AppDNA/RNA热稳定连接酶中的任一种或多种。进一步地,所述步骤(4)中,延伸时所用试剂选自DNA聚合酶、dNTPs、DNA聚合酶缓冲液、延伸引物。进一步地,所述DNA聚合酶选自ventDNA聚合酶、T7DNA聚合酶、BsuDNA聚合酶、、T4DNA聚合酶、Klenow片段、DNA聚合酶I(E.coli)、TherminatorTMDNA聚合酶、SulfolobusDNA聚合酶IV、phi29DNA聚合酶、Bst2.0DNA聚合酶、Bst2.0DNA聚合酶、BstDNA聚合酶、DeepVentR(exo–)DNA聚合酶、DeepVentRTMDNA聚合酶、VentR(exo–)DNA聚合酶、DNA聚合酶、热启动TaqDNA聚合酶、热启动TaqDNA聚合酶、TaqDNA聚合酶、热启动TaqDNA聚合酶、TaqDNA聚合酶大片段、TaqDNA聚合酶、热启动DNA聚合酶、DNA聚合酶、热启动FlexDNA聚合酶、超保真DNA聚合酶、热启动超保真DNA聚合酶、超保真DNA聚合酶等一种或几种酶的组合。进一步地,所述dNTPs可以是2.5mM的也可以是10mM的也可以是其它浓度的。所述DNA聚合酶缓冲液为对应选用的最佳匹配缓冲液。进一步地,所述延伸引物的序列与黏性末端接头的任意一条链的非黏性部分的序列全部或部分互补。所述非黏性部分是指黏性末端接头的双链部分。进一步地,所述步骤(5)中,所述双链接头的末端为平末端或黏性末端。所述双链接头的序列可以是适用于illumina测序平台的接头序列,也可以是其他双链结构的DNA。进一步地,所述黏性末端的长度为2-10nt,优选为2-5nt。进一步地,所述黏性末端可以是带有一个T的黏性末端、Y型末端或为A或T或C或G的重复序列或其它序列。例如,所述双链接头的一端可以是平末端或带有一个T的黏性末端。所述双链接头的另一端为平末端或Y型末端或黏性末端,所述黏性末端为A或T或C或G的重复序列或其它序列。进一步地,所述双链接头包括第一条链和第二条链。进一步地,所述双链接头中,第一条单链的长度为10-70nt。优选为10-30nt。再优选为10-25nt。进一步地,所述双链接头中,第一条单链的退火温度为10-100℃。优选为20-80℃。再优选为30-60℃。进一步地,所述双链接头中,第一条单链的5’端进行磷酸化修饰。进一步地,所述双链接头中,第一条单链的5’端进行间臂(spacer)修饰。进一步地,所述双链接头中,第一条单链的的5’端进行生物素化(bioten)修饰。进一步地,所述双链接头中,第一条单链的的5’端既有间臂(spacer)修饰又有生物素化(bioten)修饰。优选地,所述间臂(spacer)修饰位于所述第一条链的3’末端端序列与生物素化(bioten)修饰之间。再优选地,所述间臂(spacer)修饰与生物素之间用TEG进行连接。进一步地,所述双链接头中,第一条单链的3’端进行硫代修饰。进一步地,所述双链接头中,第二条单链的长度为10-70nt。优选为10-30nt。再优选为10-25nt。进一步地,所述双链接头中,第二条单链的的退火温度为10-100℃。优选为20-80℃。再优选为30-60℃。进一步地,所述双链接头中,第二条单链的的3’端进行间臂(spacer)修饰。优选地,采用C3Spacer或C6Spacer或SpacerC12或Spacer12或SpacerC18的修饰。再优选地,所述第二条单链的5’端采用1-10个SpacerC12或1-10个C18Spacer或1-10个C3Spacer或1-10个C6Spacer的修饰。进一步地,所述第二条单链的3’端进行生物素化(bioten)修饰。进一步地,所述二条单链的3’端既有间臂(spacer)修饰又有生物素化(bioten)修饰。优选地,所述间臂(spacer)(修饰可位于所述第二条单链的5’末端端序列与生物素化(bioten)修饰之间。再优选地,所述间臂(spacer)修饰与生物素之间用TEG进行连接。进一步地,所述第二条单链的3’端进行磷酸化修饰。进一步地,步骤(6)中,扩增时所用试剂选自DNA聚合酶、dNTPs、DNA聚合酶缓冲液、扩增引物。进一步地,所述DNA聚合酶选自ventDNA聚合酶、T7DNA聚合酶、BsuDNA聚合酶、T4DNA聚合酶、Klenow片段、DNA聚合酶I(E.coli)、TherminatorTMDNA聚合酶、SulfolobusDNA聚合酶IV、phi29DNA聚合酶、Bst2.0DNA聚合酶、Bst2.0DNA聚合酶、BstDNA聚合酶、DeepVentR(exo–)DNA聚合酶、DeepVentRTMDNA聚合酶、VentR(exo–)DNA聚合酶、DNA聚合酶、热启动TaqDNA聚合酶、热启动TaqDNA聚合酶、TaqDNA聚合酶、热启动TaqDNA聚合酶、TaqDNA聚合酶大片段、TaqDNA聚合酶、热启动DNA聚合酶、DNA聚合酶、热启动FlexDNA聚合酶、超保真DNA聚合酶、热启动超保真DNA聚合酶、超保真DNA聚合酶等中一种或几种酶的组合。进一步地,所述dNTPs可以是2.5mM的也可以是10mM的也可以是其它浓度的。所述DNA聚合酶缓冲液为对应所选聚合酶而选用的最佳匹配缓冲液。进一步地,所述扩增引物的序列与黏性末端接头的任意一条链的非黏性部分的序列全部或部分互补。所述非黏性部分是指黏性末端接头的双链部分。本发明的第二方面,提供了前述甲基化建库方法用于基因组甲基化测序或基因组甲基化位点分析中的用途。本发明的第三方面,提供了一种样本基因组甲基化测序方法,包括如下步骤:采用前述新型甲基化建库方法建立文库后,对所获得的文库进行测序。本发明的第四方面,提供一种确定单细胞样本或多细胞样本中基因组DNA甲基化位点的方法,包括如下步骤:采用前述新型甲基化建库方法建立文库后,对所获得的文库进行测序,基于测序结果,对单细胞样本或多细胞样本的基因组DNA甲基化情况进行分析,确定甲基化位点。与现有技术相比,本发明具有如下有益效果:(1)本发明提供一种新型甲基化建库方法,可适用于对于重亚硫酸盐处理的DNA样品进行单链连接、扩增或文库构建。该方案适用于单细胞、cfDNA、ctDNA、ChIpDNA、提取的线粒体DNA、提取的基因组DNA,其中提取的基因组DNA可以是活细胞或组织提取的,也可以是FFPE样本等提取的质量较差的基因组DNA。另外该方案对样品的起始量要求为6pg。(2)相比于双链连接的方案,采用本发明的新型甲基化建库方法能应用于低起始量的样品(低至6pg)。另外,该方案采用先进行重亚硫酸盐处理后再连接接头的方法,极大减少了由于药物处理造成DNA断裂导致的样品信息损失。(3)相比于随机引物扩增法,采用本发明的新型甲基化建库方法,可直接通过连接酶引入带有黏末端的双链DNA接头,因此能确保一条DNA的分子链来源于模板链的同一区域,从而实现后续比对到参考基因组时能采用更精确的双端比对的模式,增加数据的准确性。(4)相比于利用circligase进行单链连接的扩增方式,采用本发明的甲基化建库方法可通过更经济实用的诸如T4DNA连接酶进行接头连接。该酶价格便宜,且对目的DNA片段没有选择性,能更大限度的保留样品的多样性,缩小由于人为原因造成的文库偏向性。(5)采用本发明的新型甲基化建库方法进行甲基化建库可覆盖全基因组,包括富含CpG岛的启动子区域、散落的CpG位点、CHG\CHH位点;除了基因组启动子区域还包括增强子、转录起始位点、非编码区(UTR)、外显子、基因间区段(intergenic)、基因体(genebody)等,对全基因组的绝大多数胞嘧啶进行检测。(6)通过对引物的处理,可特异性地富集高甲基化的CpG岛区域或低甲基化的CpG区域或其他感兴趣的区域,应用灵活,与全基因组甲基化检测方案相比能大大减少成本。附图说明图1:本发明的甲基化建库方法中的黏性末端接头的结构示意图,NNNN为随机引物序列,图上的4个N只是展示结构,而并非对随机引物部分长度的限定。图2:展示采用本发明的甲基化建库方法对基因组DNA进行甲基化建库的大致过程,其中采用黏性末端接头连接单链DNA的步骤中,黏性末端接头中的随机引物序列NNNN,只作为结构示意,不作为限定;黏性末端接头上还可以连接生物素等修饰,后续可以与链霉亲和素等磁珠相连,进行纯化;若在连接黏性末端接头后或延伸成双链DNA后或平末端接头连接后已能满足应用,则可相应的停止后续实验,而非一定进行到指数扩增。图3:采用3%琼脂糖凝胶电泳检测单链DNA与本发明甲基化建库方法中的黏性末端接头的连接率。图4:本发明实施例1中采用本发明甲基化建库方法进行建库时,出库芯片检测结果图。具体实施方式在进一步描述本发明具体实施方式之前,应理解,本发明的保护范围不局限于下述特定的具体实施方案;还应当理解,本发明实施例中使用的术语是为了描述特定的具体实施方案,而不是为了限制本发明的保护范围;在本发明说明书和权利要求书中,除非文中另外明确指出,单数形式“一个”、“一”和“这个”包括复数形式。当实施例给出数值范围时,应理解,除非本发明另有说明,每个数值范围的两个端点以及两个端点之间任何一个数值均可选用。除非另外定义,本发明中使用的所有技术和科学术语与本
技术领域:
技术人员通常理解的意义相同。除实施例中使用的具体方法、设备、材料外,根据本
技术领域:
的技术人员对现有技术的掌握及本发明的记载,还可以使用与本发明实施例中所述的方法、设备、材料相似或等同的现有技术的任何方法、设备和材料来实现本发明。除非另外说明,本发明中所公开的实验方法、检测方法、制备方法均采用本
技术领域:
常规的分子生物学、生物化学、染色质结构和分析、分析化学、细胞培养、重组DNA技术及相关领域的常规技术。这些技术在现有文献中已有完善说明,具体可参见Sambrook等MOLECULARCLONING:ALABORATORYMANUAL,Secondedition,ColdSpringHarborLaboratoryPress,1989andThirdedition,2001;Ausubel等,CURRENTPROTOCOLSINMOLECULARBIOLOGY,JohnWiley&Sons,NewYork,1987andperiodicupdates;theseriesMETHODSINENZYMOLOGY,AcademicPress,SanDiego;Wolffe,CHROMATINSTRUCTUREANDFUNCTION,Thirdedition,AcademicPress,SanDiego,1998;METHODSINENZYMOLOGY,Vol.304,Chromatin(P.M.WassarmanandA.P.Wolffe,eds.),AcademicPress,SanDiego,1999;和METHODSINMOLECULARBIOLOGY,Vol.119,ChromatinProtocols(P.B.Becker,ed.)HumanaPress,Totowa,1999等。本发明提供了一种新型甲基化建库方法,包括如下步骤:(1)对样本的基因组DNA进行重亚硫酸盐转化;(2)对步骤(1)中重亚硫酸盐转化后的样本基因组DNA进行处理,使其维持单链DNA状态;(3)将步骤(2)中获得的单链DNA与黏性末端接头进行连接,获得一端带有接头的单链DNA;(4)对步骤(3)中获得的一端带有接头的单链DNA进行延伸,形成一端带有接头的双链DNA;(5)采用双链接头连接到步骤(4)所得一端带有接头的双链DNA的另一端,形成双端带有接头的双链DNA;(6)对步骤(5)获得的双端带有接头的双链DNA进行扩增,获得甲基化测序文库。在一个示例中,所述步骤(1)中,所述样本基因组DNA由单细胞样本裂解获得或由多细胞样本抽提获得。在一个示例中,所述步骤(1)中,若样本的基因组DNA为较长的DNA(大于500bp或500nt),则可选择先进行DNA打断后,再进行重亚硫酸盐转化;若DNA片段短则可直接进行重亚硫酸盐的转化。如需进行DNA打断操作时,可以采用超声、酶切、高温等方案进行。后续步骤同上述甲基化建库方法。在一个示例中,所述样本基因组DNA由单细胞样本裂解获得或由多细胞样本抽提获得。可以是在进行步骤(1)之前,先对单细胞样本裂解或对多细胞样本抽提,从而获得所述样本基因组DNA。也可以采用由他人利用本领域所熟知的技术已经由单细胞样本裂解获得或由多细胞样本抽提获得的样本基因组DNA。采用细胞裂解液对单细胞样本裂解或采用基因组DNA抽提试剂对多细胞样本抽提,从而获得所述样本基因组DNA。也可以采用由他人利用本领域所熟知的技术已经由单细胞样本裂解获得或由多细胞样本抽提获得的样本基因组DNA。可以采用试剂盒或者通过配置的试剂裂解一个细胞,使细胞的基因组得以释放。单个细胞的起始量一般为6pg。本技术可以单细胞为起始材料,对于细胞异质性的探索、珍惜样本的研究具有极其重要的意义。在一个示例中,所述样本的基因组DNA的质量大于等于6pg。在一个示例中,所述细胞具体为原核细胞或真核细胞。在一个示例中,所述真核细胞具体为植物细胞或动物细胞。在一个示例中,所述动物细胞具体选自组织消化的细胞、培养所得的细胞、胚胎发育早期的细胞、癌症早期的细胞、未经富集培养的微生物细胞、流式分选获得的细胞、有限稀释获得的细胞、激光捕获等方法获得的细胞中的任一种。在一个示例中,所述步骤(2)中,本领域技术人员可采用公知的技术,例如高温变性后猝冷、NaOH溶液碱变性等方案使重亚硫酸盐转化后的样本基因组DNA维持单链DNA状态。所述高温一般是指60℃-98℃。优选地为80℃-98℃。更优选地为90℃-98℃。处理时间一般为30s-10min。所述NaOH溶液为0.2~0.6MNaOH,1~1.5MNaCl。优选地为0.3~0.5MNaOH,1~1.5MNaCl。更优选地为0.4MNaOH,1.5MNaCl。本领域技术人员可以根据试剂需要来选择适合的技术,用于使重亚硫酸盐转化的样本基因组DNA维持单链DNA状态,只要不限制本发明的目的即可。在一个示例中,所述步骤(3)中,所述黏性末端接头为带有黏性末端的双链接头,所述黏性末端接头的一端为第一黏性末端,所述第一黏性末端至少包括随机序列。其中,所述随机序列用于与样本基因组DNA变性成的单链DNA通过互补配对进行连接。而所述黏性末端接头中的双链部分的序列可以适用于相应的测序平台,如illumina测序平台。如果无测序要求,则对双链部分的序列无特殊要求,只要符合双链结构即可。所述随机序列的长度可根据目标样本的多样性进行调整,若目标样本的多样性高则随机序列的长度可相对增加,若目标样本的多样性低则随机引物的长度可相对减少。通常所述随机序列的长度为2-30nt,优选的为4-20nt,更优选的为5-10nt。在一个示例中,所述黏性末端接头的另一端可以是平末端,也可以是第二黏性末端。当所述黏性末端接头的另一端为第二黏性末端时,所述第二黏性末端为任意序列或A/T/C/G寡核苷酸的重复序列,优选的为重复序列,长度优选为2-10nt,更优选为2-5bt。本领域技术人员可根据实际需要来选择第二黏性末端的序列和长度,只要不限制本发明的目的即可。在一个示例中,所述第一黏性末端包括随机序列及特殊序列。一般地,所述特殊序列为序列确定的序列。例如:当需要连接的单链DNA是通过酶切获得的带有一定末端序列的单链DNA,所述酶切可以是限制性内切酶酶切,所述限制性内切酶可以是MSPI、EcoB、EcoK、EcoRI、HindⅢ、HinfⅢ、XspI、BfaI、MaeI、TaqI、TthHB8I、HpaⅡ、HapⅡ、ApaⅠ、BamHⅠ、BanⅡ、HgiJⅡ、EcoRⅡ、MvaⅠ、BstOⅠ、BglⅠ、BglⅡ、AsuⅠ、Cfr13Ⅰ、Sau96Ⅰ、AvaⅠ、BseJⅠ、AsuⅡ、NspⅤ、BstBⅠ、Csp45Ⅰ、HgiCⅠ、BanⅠ、BstPⅠ、BstEⅡ、DraⅠ、AhaⅢ、DraⅡ等,以MSPI酶切为例,使得单链DNA带有的一定末端序列是CGG或C,那么此时,所述第一黏性末端中所包括的特殊序列是与所述一定末端序列互补的序列,亦即GCC或G。再例如,如果研究者或实验人员,对于某些目标序列具有有偏向性的富集意愿,以目标序列为CG二核苷酸序列为例,具体如CGCGCGCG,则此时,所述第一黏性末端中所包括的特殊序列是与所述目标序列互补的序列,亦即GCGCGCGC。此外,所述CG二核苷酸序列的个数可以为1-10,优选地为2-8,更优选地为2-6。在一个示例中,所述第一黏性末端的长度为2-30nt,优选的为4-20nt,更优选的为5-10nt。在一个示例中,所述黏性末端接头中,带有第一黏性末端的单链的长度为10-70nt。优选为10-30nt。再优选为10-25nt。在一个示例中,所述黏性末端接头中,带有第一黏性末端的单链的退火温度为10-100℃。优选为20-80℃。再优选为30-60℃。在一个示例中,所述第一条链的长度为10-70nt。进一步可以是10-30nt。再进一步可以是10-25nt。在一个示例中,所述第一条链的退火温度为10-100℃。进一步可以是20-80℃。再进一步可以是30-60℃。在一个示例中,所述黏性末端接头中,带有第一黏性末端的单链可进行适当的修饰,以减少引物的非特异性扩增。在一个示例中,所述黏性末端接头中,带有第一黏性末端的单链的5’端进行间臂(spacer)修饰。优选地,所述带有第一黏性末端的单链的5’端可采用连续的SpacerC12或C18Spacer或C3Spacer-CPG或C3Spacer或C9Spacer的修饰,Spacer可随意组合以增长碳链长度。在一个示例中,所述带有第一黏性末端的单链的5’端进行氨基修饰。如DMS(O)MT-Amino-Modifier-C6、Amino-Modifier-C3-TFA、Amino-Modifier-C12、Amino-Modifier-C6-TFA(trifluoroaceticacidprotectinggrouponamine)、Amino-dT、Amino-Modifier-5、Amino-Modifier-C2-dT、Amino-Modifier-C6-dT,氨基修饰可以随意组合。在一个示例中,所述带有第一黏性末端的单链的5’端既进行间臂(spacer)修饰又进行氨基修饰,间臂(spacer)修饰和氨基修饰可以随意组合。进一步地,所述带有第一黏性末端的单链的5’端采用甲基化修饰的重复A/T/C/G序列与C3Spacer或C6Spacer或Spacer12或Spacer12或SpacerC18的修饰的组合。优选地,采用甲基化修饰的重复A/T/C/G序列个数为2-8个为佳。在一个示例中,所述带有第一黏性末端的单链的3’端进行生物素化(bioten)修饰。在一个示例中,所述带有第一黏性末端的单链的3’端进行间臂(spacer)。优选地,所述带有第一黏性末端的单链的3’端可采用连续的SpacerC12或C18Spacer或C3Spacer-CPG或C3Spacer或C9Spacer的修饰,Spacer可随意组合以增长碳链长度。在一个示例中,所述带有第一黏性末端的单链的3’端进行氨基修饰。如DMS(O)MT-Amino-Modifier-C6、Amino-Modifier-C3-TFA、Amino-Modifier-C12、Amino-Modifier-C6-TFA(trifluoroaceticacidprotectinggrouponamine)、Amino-dT、Amino-Modifier-5、Amino-Modifier-C2-dT、Amino-Modifier-C6-dT,氨基修饰可以随意组合。在一个示例中,所述带有第一黏性末端的单链的3’端既进行间臂(spacer)又进行氨基修饰,间臂(spacer)和氨基修饰可以随意组合。在一个示例中,所述带有第一黏性末端的单链的3’端既有间臂(spacer)修饰又进行氨基修饰和生物素化(bioten)修饰。优选地,所述间臂(spacer)修饰和氨基修饰位于所述第一引物序列的3’末端序列与生物素化(bioten)修饰之间。再优选地,所述间臂(spacer)修饰和氨基修饰与生物素之间用TEG进行连接。在一个示例中,所述带有第一黏性末端的单链的3’端的随机序列之后还可以采用AmC3或AmC6或AmC7的氨基修饰或spacer修饰。在一个示例中,所述黏性末端接头中,不带有第一黏性末端的那条单链称为另一条单链。在一个示例中,所述黏性末端接头中,另一条单链的长度为10-70nt。优选为10-30nt。再优选为10-25nt。在一个示例中,所述黏性末端接头中,另一条单链的退火温度为10-100℃。优选为20-80℃。再优选为30-60℃。在一个示例中,所述黏性末端接头中,另一条单链的5’端进行磷酸化修饰。在一个示例中,所述黏性末端接头中,另一条单链的5’端进行间臂(spacer)修饰。优选地,采用C3Spacer或C6Spacer或SpacerC12或Spacer12或SpacerC18或C3Spacer-CPG的修饰,Spacer可随意组合以增长碳链长度。再优选地,所述黏性末端接头中,另一条单链的5’端采用1-10个SpacerC12或1-10个C18Spacer或1-10个C3Spacer或1-10个C6Spacer或1-10个C3Spacer-CPG的修饰。再优选地,所述黏性末端接头中,另一条单链的5’端采用甲基化修饰的重复A/T/C/G序列与C3Spacer或C6Spacer或Spacer12或Spacer12或SpacerC18的修饰的组合。优选地,采用甲基化修饰的重复A/T/C/G序列个数为2-8个为佳;更优选地,随机引物之后还可以采用AmC3或AmC6或AmC7的修饰或硫代修饰。在一个示例中,所述黏性末端接头中,另一条单链的5’端进行氨基修饰。如DMS(O)MT-Amino-Modifier-C6、Amino-Modifier-C3-TFA、Amino-Modifier-C12、Amino-Modifier-C6-TFA(trifluoroaceticacidprotectinggrouponamine)、Amino-dT、Amino-Modifier-5、Amino-Modifier-C2-dT、Amino-Modifier-C6-dT。在一个示例中,所述黏性末端接头中,另一条单链的3’端进行生物素化(bioten)修饰。在一个示例中,所述黏性末端接头中,另一条单链的3’端进行间臂(spacer)修饰。优选地,所述黏性末端接头中,另一条单链的5’端可采用连续的SpacerC12或C18Spacer或C3Spacer-CPG或C3Spacer或C9Spacer的修饰,Spacer可随意组合以增长碳链长度。在一个示例中,所述黏性末端接头中,另一条单链的的3’端进行氨基修饰。如DMS(O)MT-Amino-Modifier-C6、Amino-Modifier-C3-TFA、Amino-Modifier-C12、Amino-Modifier-C6-TFA(trifluoroaceticacidprotectinggrouponamine)、Amino-dT、Amino-Modifier-5、Amino-Modifier-C2-dT、Amino-Modifier-C6-dT,氨基修饰可以随意组合。在一个示例中,所述黏性末端接头中,另一条单链的3’端既进行间臂(spacer)修饰又进行氨基修饰,间臂(spacer)修饰和氨基修饰可以随意组合。在一个示例中,所述黏性末端接头中,另一条单链的3’端既有间臂(spacer)修饰又进行氨基修饰又有生物素化(bioten)修饰。优选地,所述间臂(spacer)修饰和氨基修饰位于所述第二引物序列的3’末端端序列与生物素化(bioten)修饰之间。再优选地,所述间臂(spacer)和氨基修饰与生物素之间用TEG进行连接。本发明一些实施例中所列举的是适用于illumina测序平台的接头序列,仅用于示意,并不局限于这些具体的序列。实施例中所列举的修饰方式也仅用于示意。可根据实际需要进行其他修饰。在一个示例中,所述步骤(3)中,所述黏性末端接头通过连接酶连接到所述单链DNA上。在一个示例中,所述连接酶选自HiFiTaqDNA连接酶、T4RNA连接酶2、连接酶、9°NTMDNA连接酶、TaqDNA连接酶、T7DNA连接酶、T3DNA连接酶、电转连接酶、平末端/TA连接酶预混液、瞬时黏性连接酶预混液、T4DNA连接酶、T4RNA连接酶、CircligasessDNAligase、5′AppDNA/RNA热稳定连接酶中的任一种或多种。在一个示例中,所述步骤(4)中,延伸时所用试剂选自DNA聚合酶、dNTPs、DNA聚合酶缓冲液、延伸引物。在一个示例中,所述DNA聚合酶选自ventDNA聚合酶、T7DNA聚合酶、BsuDNA聚合酶、、T4DNA聚合酶、Klenow片段、DNA聚合酶I(E.coli)、TherminatorTMDNA聚合酶、SulfolobusDNA聚合酶IV、phi29DNA聚合酶、Bst2.0DNA聚合酶、Bst2.0DNA聚合酶、BstDNA聚合酶、DeepVentR(exo–)DNA聚合酶、DeepVentRTMDNA聚合酶、VentR(exo–)DNA聚合酶、DNA聚合酶、热启动TaqDNA聚合酶、热启动TaqDNA聚合酶、TaqDNA聚合酶、热启动TaqDNA聚合酶、TaqDNA聚合酶大片段、TaqDNA聚合酶、热启动DNA聚合酶、DNA聚合酶、热启动FlexDNA聚合酶、超保真DNA聚合酶、热启动超保真DNA聚合酶、超保真DNA聚合酶等一种或几种酶的组合。在一个示例中,所述DNA聚合酶缓冲液为对应选用的最佳匹配缓冲液。在一个示例中,所述延伸引物的序列与黏性末端接头的任意一条链的非黏性部分的序列全部或部分互补。所述非黏性部分是指黏性末端接头的双链部分。在一个示例中,所述步骤(5)中,所述双链接头的末端为平末端或黏性末端。所述双链接头的序列可以是适用于illumina测序平台的接头序列,也可以是其他双链结构的DNA。在一个示例中,所述黏性末端的长度为2-10nt,优选为2-5nt。在一个示例中,所述黏性末端可以是带有一个T的黏性末端、Y型末端或为A或T或C或G的重复序列或其它序列。本领域技术人员可根据实际需要来选择所述黏性末端的序列,只要不限制本发明的目的即可。例如,所述双链接头的一端可以是平末端或带有一个T的黏性末端。所述双链接头的另一端为平末端或Y型末端或黏性末端,所述黏性末端为A或T或C或G的重复序列或其它序列。在一个示例中,所述双链接头包括第一条链和第二条链。在一个示例中,所述双链接头中,第一条单链的长度为10-70nt。优选为10-30nt。再优选为10-25nt。在一个示例中,所述双链接头中,第一条单链的退火温度为10-100℃。优选为20-80℃。再优选为30-60℃。在一个示例中,所述双链接头中,第一条单链的5’端进行磷酸化修饰。在一个示例中,所述双链接头中,第一条单链的5’端进行间臂(spacer)修饰。在一个示例中,所述双链接头中,第一条单链的的5’端进行生物素化(bioten)修饰。在一个示例中,所述双链接头中,第一条单链的的5’端既有间臂(spacer)修饰又有生物素化(bioten)修饰。优选地,所述间臂(spacer)修饰位于所述第一条链的3’末端端序列与生物素化(bioten)修饰之间。再优选地,所述间臂(spacer)修饰与生物素之间用TEG进行连接。在一个示例中,所述双链接头中,第一条单链的3’端进行磷酸化修饰。在一个示例中,所述双链接头中,第一条单链的3’端进行硫代修饰。在一个示例中,所述双链接头中,第二条单链的长度为10-70nt。优选为10-30nt。再优选为10-25nt。在一个示例中,所述双链接头中,第二条单链的的退火温度为10-100℃。优选为20-80℃。再优选为30-60℃。在一个示例中,所述双链接头中,第二条单链的5’端进行磷酸化修饰。在一个示例中,所述双链接头中,第二条单链的的3’端进行间臂(spacer)修饰。优选地,采用C3Spacer或C6Spacer或SpacerC12或Spacer12或SpacerC18的修饰。再优选地,所述第二条单链的5’端采用1-10个SpacerC12或1-10个C18Spacer或1-10个C3Spacer或1-10个C6Spacer的修饰。再优选地,所述第二条单链的5’端采用甲基化修饰的重复A/T/C/G序列与C3Spacer或C6Spacer或Spacer12或Spacer12或SpacerC18的修饰的组合。优选地,采用甲基化修饰的重复A/T/C/G序列个数为2-8个为佳。在一个示例中,所述第二条单链的3’端进行生物素化(bioten)修饰。在一个示例中,所述二条单链的3’端既有间臂(spacer)修饰又有生物素化(bioten)修饰。在一个示例中,所述间臂(spacer)修饰可位于所述第二条单链的5’末端端序列与生物素化(bioten)修饰之间。再优选地,所述间臂(spacer)修饰与生物素之间用TEG进行连接。在一个示例中,所述第二条单链的3’端进行生物素化硫代修饰。在一个示例中,所述第二条单链的3’端进行磷酸化修饰。在一个示例中,所述步骤(6)中,扩增时所用试剂选自DNA聚合酶、dNTPs、DNA聚合酶缓冲液、扩增引物。在一个示例中,所述DNA聚合酶选自ventDNA聚合酶、T7DNA聚合酶、BsuDNA聚合酶、T4DNA聚合酶、Klenow片段、DNA聚合酶I(E.coli)、TherminatorTMDNA聚合酶、SulfolobusDNA聚合酶IV、phi29DNA聚合酶、Bst2.0DNA聚合酶、Bst2.0DNA聚合酶、BstDNA聚合酶、DeepVentR(exo–)DNA聚合酶、DeepVentRTMDNA聚合酶、VentR(exo–)DNA聚合酶、DNA聚合酶、热启动TaqDNA聚合酶、热启动TaqDNA聚合酶、TaqDNA聚合酶、热启动TaqDNA聚合酶、TaqDNA聚合酶大片段、TaqDNA聚合酶、热启动DNA聚合酶、DNA聚合酶、热启动FlexDNA聚合酶、超保真DNA聚合酶、热启动超保真DNA聚合酶、超保真DNA聚合酶等中一种或几种酶的组合。所述dNTPs可以是2.5mM的也可以是10mM的也可以是其它浓度的。本领域技术人员可根据实际需要来选择dNTPs的浓度,只要不限制本发明的目的即可。在一个示例中,所述DNA聚合酶缓冲液为对应选用的最佳匹配缓冲液。在一个示例中,所述扩增引物的序列与黏性末端接头的任意一条链的非黏性部分的序列全部或部分互补。所述非黏性部分是指黏性末端接头的双链部分。本发明的甲基化建库方法可以用于用于基因组甲基化测序或基因组甲基化位点分析。因此,本发明还提供了一种样本基因组甲基化测序方法,包括如下步骤:采用前述新型甲基化建库方法建立文库后,对所获得的文库进行测序。进一步地,所述测序可以是一代测序、二代测序或三代测序。进一步地,所述测序的平台可以是illumina测序平台。本领域技术人员还可采用前述新型甲基化建库方法建立文库后,对所获得的文库进行测序,基于测序结果,对单细胞样本或多细胞样本的基因组DNA甲基化情况进行分析,确定甲基化位点。采用本发明的新型甲基化建库方法进行甲基化文库建立,先进行重亚硫酸盐处理后再连接带有黏末端的双链接头,极大减少了由于药物处理造成DNA断裂导致的样品信息损失。重亚硫酸盐可以将未甲基化的胞嘧啶(C)转化为为尿嘧啶(U)。重亚硫酸盐的转化不仅可以是zymoresearch的试剂盒,也可以是其他具有类似功效的试剂盒或自己配置的试剂。相比于现有技术中的随机引物扩增法,采用本发明的新型甲基化建库方法进行甲基化文库建立,可直接采用连接酶引入带有黏末端的双链DNA接头序列,因此,能确保一条DNA的分子链来源于模板链的同一区域,从而实现后续比对到参考基因组时能采用更精确的双端比对的模式,增加数据的准确性。相比于现有技术中利用circligase进行单链连接的扩增方式,采用本发明的新型甲基化建库方法进行甲基化文库建立,可采用更经济实用的普通连接酶例如T4DNA连接酶进行接头连接。这些普通的连接酶价格便宜,且对目的DNA片段没有选择性,能更大限度的保留样品的多样性,缩小由于人为原因造成的文库偏向性。采用本发明的新型甲基化建库方法进行甲基化建库,操作灵活,可根据需要采用特异性带有黏末端的双链接头直接连接单链DNA,经过扩增得到的扩增子适用于后续的一系列应用,如可以用于构建一代测序文库,也可以用于构建二代测序文库,也可以构建三代测序文库,也可以用于qPCR模板的建立、还可以用于短片段DNA的克隆等。使用者可以根据构建的测序文库的类别,采用相应的PCR进行扩增,以构建不同的测序文库。需要说明的是,采用本发明的新型甲基化建库方法进行甲基化建库的起始样本可以为一个细胞的基因组,仅用于说明而非限定。容易理解的是,所述起始样本也可以是多个细胞的基因组。采用本发明的新型甲基化建库方法进行甲基化建库可覆盖全基因组,包括富含CpG岛的启动子区域、散落的CpG位点、CHG\CHH位点;除了基因组启动子区域还包括增强子、转录起始位点、非编码区(UTR)、外显子、基因间区段(intergenic)、基因体(genebody)等,对全基因组的绝大多数胞嘧啶进行检测。另外通过对引物的处理,还可特异性地富集高甲基化的CpG岛区域或低甲基化的CpG区域或其他感兴趣的区域,应用灵活,与全基因组甲基化检测方案相比能大大减少成本。下文以具体实施例1对本发明实施例的技术方案进行更具体地说明。实施例1一、试验制备(一)、甲基化建库方法中要用到的黏性末端接头合成:所述黏性末端接头为带有黏性末端的双链接头,所述黏性末端接头的一端为第一黏性末端,所述第一黏性末端至少包括随机序列。1、作为示例,采用Primer1和Primer2合成黏性末端接头,Primer1:5’-AGATCGGAAG-3’-bioten(SEQIDNO.1);Primer2:5’-CTTCCGATCTNNNNNN-3’(SEQIDNO.2);其中,NNNNNN表示随机序列,通常所述随机序列的长度为2-30nt,优选的为4-20nt,更优选的为5-10nt。2、按下表1加入配置反应液:表1试剂体积(ul)Primer1(10μM)1Primer2(10μM)1TEbuffer75MNaCl1总体积10其中反应中采用的TEbuffer也可以被水、Tris-Hcl(0.05mM,PH7.0-8.0)等具有相似功能的缓冲液所替代。3、按下表2进行反应:表2温度时间循环数95℃60s195℃-16℃0.5℃/s116℃forever1获得黏性末端接头:5’-CTTCCGATCT-NNNNNN-3’bioten-3’-GAAGGCTAGA-5’。可转存于-20℃保存。亦即,示例性地,Primer2和Primer1一起形成了黏性末端接头,所述黏性末端接头为带有黏性末端的双链接头,所述黏性末端接头的一端为第一黏性末端,所述第一黏性末端包括随机序列NNNNNN,通常所述随机序列NNNNNN的长度为2-30nt,优选的为4-20nt,更优选的为5-10nt。若将带有第一黏性末端的单链称为第一条链,不带有第一黏性末端的单链称为第二条链,则第二条链的3’进行了bioten修饰。在此示例中,所述黏性末端接头的另一端为平末端。表2中的反应条件只是作为示例,也可以采用类似的方案进行,总体原则为先高温变性后缓慢退火形成互补的DNA双链。(二)、甲基化建库方法中要用到的双链接头的准备1、作为示例,双链接头由Primer3和Primer4制备而成,,其中一个末端为黏性末端。另一个末端为平末端。Primer3:5’-CTTCCGATC-3’(SEQIDNO.3);Primer4:5’-GATCGGAAGAGCGTCGTGTAGGG-3’(SEQIDNO.4);Primer3在3’端优选地采用磷酸化修饰的C,更优选地可以采用双脱氧核糖核酸的C进行引物的合成;Primer4可以选择进行硫代修饰或不进行硫代修饰,优选的为进行硫代修饰,更优选的为硫代修饰3’端的3个碱基;primer4的5’端优选地采用磷酸化修饰。2、按下表3加入配置反应液:表3试剂体积(ul)Primer3(10μM)5Primer4(10μM)5TEbuffer95MNaCl1总体积203、按下表4进行反应:表4温度时间循环数95℃60s195℃-16℃0.5℃/s116℃forever1获得双链接头可转存于-20℃保存。上表5中的反应条件只是作为示例,也可以采用类似的方案进行,总体原则为先高温变性后缓慢退火是互补的DNA双链结构。(三)甲基化建库方法中要用到的重亚硫酸盐转化试剂、连接酶、DNA延伸试剂、DNA扩增试剂可通过商业购买途径获得。具体地,所述DNA延伸试剂包括DNA聚合酶、dNTPs、DNA聚合酶缓冲液、延伸引物。所述延伸引物的序列与黏性末端接头的任意一条链的非黏性部分的序列全部或部分互补。所述非黏性部分是指黏性末端接头的双链部分。作为示例,所述延伸引物使用:Primer5:5’-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATC-3’(SEQIDNO.5)。所述DNA扩增试剂包括DNA聚合酶、dNTPs、DNA聚合酶缓冲液、扩增引物。所述扩增引物的序列与黏性末端接头的任意一条链的非黏性部分的序列全部或部分互补。所述非黏性部分是指黏性末端接头的双链部分。作为示例,所述扩增引物使用:Primer6:5’-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT-3’(SEQIDNO.6);Primer7:5’-CAAGCAGAAGACGGCATACGAGATtgccgaGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3’(SEQIDNO.7)。二、采用本发明的甲基化建库方法对样本基因组DNA进行甲基化文库建立(一)细胞裂解或抽提获得目的DNA(亦即样本基因组DNA)a)按表5的体系建立裂解体系:表5成分体积(μl)Tris-EDTA(500mMTris,50mMEDTA)0.051MKCl0.0510%(vol/vol)TritonX-1000.0510mg/ml蛋白酶0.25无核酸水补至4b)涡旋混匀裂解体系。c)6000g,4℃,离心1min。确保所有样品被收集在PCR管底部。d)将裂解体系转移到含有单细胞的PCR管中,共5μl。e)6000g,4℃,离心1min。f)PCR仪设置50℃,30分钟到过夜不等,可以根据样本的不同需要进行具体调整。裂解细胞;75℃,30min,4℃保温。g)6000g,4℃,离心1min。h)立即转移到冰上。样品也可以由抽提获得。(二)重亚硫酸盐转化目的DNA(亦即样本基因组DNA)可以使用zymoresearch等公司的商业化试剂盒,也可以是自己配置的试剂进行重亚硫酸盐转化。以下以zymoresearch的试剂盒进行具体举例说明。a)20μl样品(6pg-2μg),瞬时离心以将样品收集于PCR管底部。b)加入130μlCTConversionReagent吹打混匀或轻弹混匀,瞬时离心。c)严格按照试剂盒的说明书进行后续操作需要说明的是在转化后的回收过程中可以加入糖原、carrierRNA、重复序列DNA或不会影响后续扩增或分析的核酸及化学物质提高回收效率,也可以采用其他DNA回收试剂盒或其他DNA回收方案替换。(三)对样本的基因组DNA进行去磷酸化,防止样本的基因组DNA之间的自连,这一步骤为可选步骤具体地,按下表6加入配置反应液:表6试剂体积(ul)10xT4DNAligationbuffer41%Tween201FastAP(1U/μl)1样本的基因组DNA(6pg-100ng)14总体积20这里的样本的基因组可以是采用细胞裂解液对单细胞样本裂解或采用基因组DNA抽提试剂对多细胞样本抽提获得。也可以采用由他人利用本领域所熟知的技术已经由单细胞样本裂解获得或由多细胞样本抽提获得的样本基因组DNA。具体地,将样品轻弹混匀后,按下表7进行反应:表7温度时间37℃30min95℃5min(四)采用本领域技术人员公知的技术如高温变性后猝冷的方案使重亚硫酸盐转化后的样本基因组DNA维持单链DNA状态具体的将样品70-98℃加热3min后将样品立即转移到冰上,优选地为80-95℃。(五)单链DNA通过连接酶与黏性末端接头进行连接,获得一端带有接头的单链DNA具体地,按下表8加入配置反应液:表8试剂体积(ul)50%PEG-40004100mMATP0.1本实施例中获得的黏性末端接头1T4DNA连接酶1无核酸水13.9总体积20轻弹混匀,总体积40ul。然后,按下表9进行反应:表9温度时间循环数37℃30min195℃5min14℃forever1可将样品于-20℃保存,或直接进行下一步反应。如图3所示,采用3%琼脂糖凝胶电泳检测单链DNA与黏性末端接头的连接率。其中,阴对:未加连接酶的阴性对照,10ng;1:表示1号样品,10ng;2:表示2号样品,50ng;3:表示3号样品,100ng。S+A:连接成功的样品加上接头,产物长度大于样品故在胶上显示跑的较慢;S:样品,未连接接头,分子量较小,故在胶上显示跑的较快;A:接头。从图3可以看出,在不同样品起始量时,都有较好的连接结果,在样品起始量低时基本可完全连接上接头。在样品量升高时,有部分样品没能连接成功,但是由于接头已被耗尽,故提高接头含量可提高量样品的连接率。DNA样品在与黏性末端接头连接时,可以不用FastAP进行处理,优选的为利用FastAP进行处理。反应中采用的1%Tween20可以被同浓度的NP40、tritonx-100所替代;T4DNA连接酶可以由其他具有双链或单链连接酶活性的酶所替代,优选地有HiFiTaqDNA连接酶、T4RNA连接酶2、连接酶、9°NTMDNA连接酶、TaqDNA连接酶、T7DNA连接酶、T3DNA连接酶、电转连接酶、平末端/TA连接酶预混液、瞬时黏性连接酶预混液、T4RNA连接酶、CircligasessDNAligase、5′AppDNA/RNA热稳定连接酶。优选地使用T4DNA连接酶。(六)、磁珠捕获一端带有接头的单链DNA,此步骤可视具体情况选择,为可选步骤1、将链霉亲和素修饰的磁珠,在室温至少平衡30分钟,涡旋混匀。2、取20ul的beads到1.5ml的EP管中(20ul/样品)。3、Beads用500μl的1xBindingbuffer(1MNaCl,10mMTris-HCl(pH8.0),1mMEDTA(pH8.0),0.05%Tween-20,0.5%SDS)洗涤2次。4、将beads重悬于100ul的1xBindingbuffer。5、将样品(40ul)在95℃加热1min,迅速转移到冰上,放置至少2-5min。6、将样品与洗涤完成的beads混匀。7、在室温孵育40min,并不断混匀。8、瞬离,将EP管放置在磁力架上,去除上请。9、加入200μl的washbufferI(0.1MNaCl,10mMTris-HCl(pH8.0),1mMEDTA(pH8.0),0.05%Tween-20,0.5%SDS),涡旋8s使beads重悬。10、瞬离,将EP管转移到磁力架上,去除上清。其中链霉亲和素修饰的磁珠可以是DynabeadsTMM-280Streptavidin、DynabeadsTMMyOneTMStreptavidinC1、DynabeadsTMM-270Streptavidin等商业产品,也可以是自行偶联的链霉亲和素修饰的类似磁珠样品。(七)、对一端带有接头的单链DNA进行延伸,形成一端带有接头的双链DNA具体地,按下表10加入配置反应液:表10试剂体积(ul)10xKlenowreactionbuffer510mMdNTPs0.41%Tween-202.5Primer5(10μM)1水39.1总体积48Primer5:5’-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATC-3’(SEQIDNO.5)。可选的,primer5可进行硫代修饰,也可不进行硫代修饰,优选的进行硫代修饰,更优选地为整条引物进行硫代修饰,更优选地为3端的10个碱基进行硫代修饰;更优选的为3端的5个碱基进行硫代修饰;更优选地为3端的个碱基进行硫代修饰。1.将beads转移到磁力架上,去除上请。2.加入上表10中的混合物,涡旋使beads充分混匀。3.将样品转移到95℃,2min。4.迅速转移到冰上,至少放置2-5min。5.将样品在室温转移到EP管中。6.加入2ul的Klenowfragment(10U/μl),混匀,将beads重悬。7.25℃孵育5min,每60s混匀2s,1500rpm。8.35℃孵育25min,每60s混匀2s,1500rpm。其中Klenowfragment也可以被其他的聚合酶所替代,如:ventDNA聚合酶、T7DNA聚合酶、BsuDNA聚合酶,大片段、T4DNA聚合酶、Klenow片段、DNA聚合酶I(E.coli)、TherminatorTMDNA聚合酶、SulfolobusDNA聚合酶IV、phi29DNA聚合酶、Bst2.0DNA聚合酶、Bst2.0DNA聚合酶、BstDNA聚合酶,全长、BstDNA聚合酶,DeepVentR(exo–)DNA聚合酶、DeepVentRTMDNA聚合酶、VentR(exo–)DNA聚合酶、DNA聚合酶、热启动TaqDNA聚合酶、热启动TaqDNA聚合酶、TaqDNA聚合酶、热启动TaqDNA聚合酶、TaqDNA聚合酶大片段、TaqDNA聚合酶、热启动DNA聚合酶、DNA聚合酶、热启动FlexDNA聚合酶、超保真DNA聚合酶、热启动超保真DNA聚合酶、超保真DNA聚合酶等一种或几种酶的组合。上述反应体系只用于实例展示,而不用于限定。(八)、扩增后清洗,为可选步骤1.样品瞬离,转移到磁力架上,去除上清。2.加入200μl的washbufferI(0.1MNaCl,10mMTris-HCl(pH8.0),1mMEDTA(pH8.0),0.05%Tween-20,0.5%SDS),涡旋8s使beads重悬。3.将样品混匀,瞬离,转移到磁力架上,去除上清。4.加入100μl的washbufferII(0.5xSSC,PH7.0)。5.将样品室温孵育3min。6.瞬离,转移到磁力架上,去除上请。7.加入200μl的washbufferI(0.1MNaCl,10mMTris-HCl(pH8.0),1mMEDTA(pH8.0),0.05%Tween-20,0.5%SDS),涡旋8s使beads重悬。上述清洗方案只用与实例展示而不用于限定。(九)、采用双链接头连接到一端带有接头的双链DNA的另一端,形成双端带有接头的双链DNA按下表10加入配置反应液:表11试剂体积(ul)10xT4DNAligasebuffer550%PEG-40005双链接头(10uM)21%Tween-202T4DNAligase(5U/μl)2水34总体积501.将beads去上清,用上表10中的mix混匀。2.将样品在25℃孵育1h,孵育时注意混匀。T4DNA连接酶可以由其他具有双链或单链连接酶活性的酶所替代,优选地有HiFiTaqDNA连接酶、T4RNA连接酶2、连接酶、9°NTMDNA连接酶、TaqDNA连接酶、T7DNA连接酶、T3DNA连接酶、电转连接酶、平末端/TA连接酶预混液、瞬时黏性连接酶预混液、T4DNA连接酶、T4RNA连接酶、CircligasessDNAligase、5′AppDNA/RNA热稳定连接酶、T4RNALigase等一种或几种酶的组合。优选地使用T4DNA连接酶。(十)、连接后洗涤及样品洗脱1.将样品瞬离,转移到磁力架上,去除上请。2.加入200μl的washbufferI(0.1MNaCl,10mMTris-HCl(pH8.0),1mMEDTA(pH8.0),0.05%Tween-20,0.5%SDS)。3.将样品混匀。4.瞬离,转移到磁力架上,去除上清。5.加入100μl的Stringencywashbuffer(0.1xSSCand0.1%SDS)。6.将样品在45℃,孵育3min。7.瞬离,转移到磁力架上,去除上请。8.加入200μl的washbufferI(0.1MNaCl,10mMTris-HCl(pH8.0),1mMEDTA(pH8.0),0.05%Tween-20,0.5%SDS),涡旋8s使beads重悬。9.将样品转移到混匀,转移到磁力架上,去除上请。10.加入50μl的无核酸水。11.将样品在室温孵育10min后,95℃加热2min。12.将样品转移到磁力架上,将上清转移到干净的低吸付的EP管中。上述双链接头连接方案只用于实例说明,而不用于限定。(十一)、采用DNA扩增试剂对双端带有接头的双链DNA进行扩增,获得测序文库(可选步骤)按下表12混合pcr扩增原料:表12样品体积(ul)KAPAHiFiHotStartReadyMix(2x)25Primer6(10μM)1Primer7(10μM)1NFwater23总体积50将洗脱的样品或直接带着磁珠进行下列反应(每个样品可做2个或多个平行的PCR扩增反应,可增加样品的产量)。混匀样品,按下表13进行扩增:表13Primer6:5’-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT-3’(SEQIDNO.6);Primer7:5’-CAAGCAGAAGACGGCATACGAGATtgccgaGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3’(SEQIDNO.7);其中下划线部分为index序列,可以替换为其他的index序列,具体可参考illumina测序仪器适用的index序列。如果不需要进行文库测序,只是进行扩增,则可利用以下引物进行:Primer8:5’-CACTCTTTCCCTACACGACGCTCTTCCGATCT-3’(SEQIDNO.8);Primer9:5’-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATC-3’(SEQIDNO.9);其中可选取Primer8或Primer9的全长引物进行扩增,也可以是选取其中的一部分作为PCR引物,优选的选用全长引物进行扩增。其中KAPAHiFiHotStartReadyMix(2x)也可以由其他聚合酶所替代,如ventDNA聚合酶、T7DNA聚合酶、BsuDNA聚合酶,大片段、T4DNA聚合酶、Klenow大片段、DNA聚合酶I(E.coli)、TherminatorTMDNA聚合酶、SulfolobusDNA聚合酶IV、phi29DNA聚合酶、Bst2.0DNA聚合酶、Bst2.0DNA聚合酶、BstDNA聚合酶,全长、BstDNA聚合酶,DeepVentR(exo–)DNA聚合酶、DeepVentRTMDNA聚合酶、VentR(exo–)DNA聚合酶、DNA聚合酶、热启动TaqDNA聚合酶、热启动TaqDNA聚合酶、TaqDNA聚合酶、热启动TaqDNA聚合酶、TaqDNA聚合酶大片段、TaqDNA聚合酶、热启动DNA聚合酶、DNA聚合酶、热启动FlexDNA聚合酶、超保真DNA聚合酶、热启动超保真DNA聚合酶、超保真DNA聚合酶、KAPAHiFiUracil+ReadyMix(2x)、PfuTurboCx热启动DNA聚合酶等一种或几种酶的组合。上述反应只用于实例展示,而不用于限定。(十二)、AmpureXPbeads纯化样品1.将AMPureXPBeadsDNA纯化磁珠在室温平衡至少30min。2.取40μlDNA纯化磁珠AgencourtAMPureXPBeads到50μl的样品中,涡旋混匀。若同一样品进行了平行的PCR反应,可将多个样品混合成一管。其中DNA纯化磁珠AgencourtAMPureXPBeads的用量可更具需要进行调整,优选的为40-50ul。3.室温放置5min,使样品与DNA充分结合。4.将样品转移到磁力架上,待样品澄清后弃去上清。5.用新鲜的200ul80%(vol/vol)的乙醇(现配现用),清洗beads,可上下颠倒,转动EP管,使洗涤充分。6.弃去上清。7.用新鲜的200ul80%(vol/vol)的乙醇(现配现用),清洗beads,可上下颠倒,转动EP管,使洗涤充分。弃去上清。8.瞬离一次,待样品澄清后弃去残留乙醇。9.室温开盖等待,使乙醇充分挥发,至表面出现小裂痕。(不可过干或有乙醇残留)。10.加入适量的无核酸水,与样品充分混匀。11.在室温孵育至少2min。12.将上清转移至干净的NF的EP管中,余2ul进行定量与质检。其中DNA纯化磁珠可以是AMPureXPBeads可以由等其他具有相同功效的磁珠替代,除了可以用磁珠纯化样品,也可以采用具有相同功效的试剂盒或方案替代。本实施例文库出库芯片检测结果图如图4所示,安捷伦2100高灵敏芯片检测文库出库后的片段大小,X轴为片段大小,Y轴为荧光强度。该文库的大小为100bp-400bp,其中分布在220bp左右的片段达到峰值。下机数据质控分析,如表14所示:表14如果某碱基质量值为20(Q20),则表示该碱基测序出错的概率为0.01。如果某碱基质量值为30(Q30),则表示该碱基测序出错的概率为0.001。Map_rate:能回帖到基因组的数据占下机数据的比例。Sample1、Sample2、Sample3、Sample4、Sample5、Sample6、Sample7、Sample8的样品来源为FFPE样本提取的DNA,平均分为8份,每份100ng。Sample1、Sample2、Sample3、Sample4采用常规illumina双链连接的方案进行建库(具体采用试剂盒如:IlluminaSamplePreparationKits;CustomAmpliconorAmplicon–CancerPanel;TargetedRNAExpression;HTSamplePrepKits;v1/v2/LTSamplePrepKits;SyntheticLong-ReadDNALibraryPrepKits;SmallRNASamplePrepKits;DNASamplePrepKit;MultiplexingSamplePrepOligoOnlyKit;v1andv1.5SmallRNAKits;SamplePreparationKits),而Sample5、Sample6、SSample7、ample8、采用本发明中的新型甲基化建库试剂盒通过单链DNA与黏性末端接头连接的方式进行建库。从表14中可知两种方案,采用本发明中的新型甲基化建库试剂盒通过单链DNA与黏性末端接头连接的方式进行建库的方式在样本的出库产量、测序质量、双端比对效率明显优于常规illumina双链连接的方案,illumina建库试剂盒基本不能对如此少量的DNA样本进行建库。上述实施例仅例示性说明本发明的原理及其功效,而非用于限制本发明。任何熟悉此技术的人士皆可在不违背本发明的精神及范畴下,对上述实施例进行修饰或改变。因此,举凡所属
技术领域:
中具有通常知识者在未脱离本发明所揭示的精神与技术思想下所完成的一切等效修饰或改变,仍应由本发明的权利要求所涵盖。序列表<110>上海美吉生物医药科技有限公司<120>一种新型甲基化建库方法及其应用<130>176455<160>9<170>SIPOSequenceListing1.0<210>1<211>10<212>DNA<213>人工序列(ArtificialSequence)<400>1agatcggaag10<210>2<211>16<212>DNA<213>人工序列(ArtificialSequence)<400>2cttccgatctnnnnnn16<210>3<211>9<212>DNA<213>人工序列(ArtificialSequence)<400>3cttccgatc9<210>4<211>23<212>DNA<213>人工序列(ArtificialSequence)<400>4gatcggaagagcgtcgtgtaggg23<210>5<211>33<212>DNA<213>人工序列(ArtificialSequence)<400>5gtgactggagttcagacgtgtgctcttccgatc33<210>6<211>58<212>DNA<213>人工序列(ArtificialSequence)<400>6aatgatacggcgaccaccgagatctacactctttccctacacgacgctcttccgatct58<210>7<211>64<212>DNA<213>人工序列(ArtificialSequence)<400>7caagcagaagacggcatacgagattgccgagtgactggagttcagacgtgtgctcttccg60atct64<210>8<211>32<212>DNA<213>人工序列(ArtificialSequence)<400>8cactctttccctacacgacgctcttccgatct32<210>9<211>33<212>DNA<213>人工序列(ArtificialSequence)<400>9gtgactggagttcagacgtgtgctcttccgatc33当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1