一种2-苄基-3,4-二氢-2H-异喹啉-1-酮衍生物的制备方法与流程
本发明属于有机化学领域,涉及一种异喹啉-1-酮衍生物,具体来说是一种2-苄基-3,4-二氢-2h-异喹啉-1-酮衍生物的制备方法。
背景技术:
2-苄基-3,4-二氢-2h-异喹啉-1-酮衍生物属于天然杂环化合物,具有极重要的生理活性和生物活性,是非常重要的生物活性药物。因此研究其更有效的制备方法非常受研究人员的重视。目前,关于异喹啉类功能化物质的报道均是采用多步法,利用带有或不带有保护基团的异喹啉反应得到相应产物后,再去保护基得到反应中间体,然后和苯甲醛反应得到最终产物。这种方法需要至少三步反应,过程繁琐,产率低。因此,一种高效的异喹啉功能化反应是非常有应用前景的。
技术实现要素:
针对现有技术中的上述技术问题,本发明提供了一种2-苄基-3,4-二氢-2h-异喹啉-1-酮衍生物的制备方法,所述的这种2-苄基-3,4-二氢-2h-异喹啉-1-酮衍生物的制备方法要解决现有技术中制备2-苄基-3,4-二氢-2h-异喹啉-1-酮衍生物的方法工艺复杂、产率低的技术问题。
本发明提供了一种2-苄基-3,4-二氢-2h-异喹啉-1-酮衍生物的制备方法,以r1取代的苯甲醛、苄基叠氮和四氢异喹啉为原料,在有机溶剂中,敞口条件下,加入分子筛,在铜催化剂和添加剂存在的条件下,80-100℃的温度下反应2-5h,将所得的反应液在压力为0.02-0.04mpa的条件下加压过滤3-5min,所得的滤液先控制温度为30-45℃进行浓缩3-5min,所得的浓缩液用旋转蒸发仪控制温度为25-45℃下进行旋转蒸发4-8min,然后在洗脱液下进行柱层析分离,所分离出来的产物即为2-苄基-3,4-二氢-2h-异喹啉-1-酮衍生物;
所述的r1取代的苯甲醛中,r1为供电子基或吸电子基,所述的供电子基为
h或甲基;所述的吸电子基为溴或氯;
所述的铜催化剂为cucl2、cubr、cui、cucl、cu(no3)2·3h2o或cubr2;
所述的添加剂为冰醋酸、苯甲酸、乙酸、苯乙酸或三氟乙酸;
所述的r1取代的苯甲醛、苄基叠氮、四氢异喹啉的摩尔比为1~1.4:1~1.4:1~1.4;
所述的铜催化剂和苄基叠氮的摩尔比为5~10:100;
所述的添加剂和苄基叠氮的摩尔比为5~10:100;
所述的苄基叠氮和有机溶剂的物料比为0.5mmol:2-3ml。
进一步的,所述的有机溶剂为甲苯。
进一步的,所述的r1取代的苯甲醛为苯甲醛、2-氯苯甲醛、2-溴苯甲醛、2-碘苯甲醛、3-氯苯甲醛、2,4-二氯苯甲醛、2-溴-4-氯苯甲醛、2,4-二氯苯甲醛、2-溴-5-氟苯甲醛、4-甲基苯甲醛、4-乙基苯甲醛、2-溴-4-甲基苯甲醛;所述的铜催化剂为cucl2;所述的添加剂为冰乙酸。
本发明的制备过程的反应方程式如下所示:
即采用r1取代的苯甲醛,四氢异喹啉和苄基叠氮为原料,在有机溶剂甲苯中,敞口条件下,以金属铜为催化剂,在添加剂(作为反应体系的酸)存在的条件下,控制温度为80-100℃进行反应2-5h,反应进程用薄层色谱进行跟踪;上述反应结束后,将所得的反应液进行过滤,所得的滤液先控制温度为30-45℃进行浓缩3-5min,所得的浓缩液用旋转蒸发仪控制温度为25-45℃下旋转蒸发4-8min,再用石油醚/乙酸乙酯=10:1条件下进行柱层析分离,所分离出来的产物为r1取代的2-苄基-3,4-二氢-2h-异喹啉-1-酮,即为2-苄基-3,4-二氢-2h-异喹啉-1-酮衍生物。
本发明和已有技术相比,其技术进步是显著的。本发明采用四氢异喹啉、苯甲醛类化合物和苄基叠氮一步反应法,得到带有不同有取代基的异喹啉衍生物。本发明由于采用“一锅法”进行反应制备,因此具有反应条件简单,不需要进行中间分离的优点,从而解决了现有技术中过程繁琐,产率低的问题,本发明的制备方法最终产物的产率最高可达85%,同时大大提高了反应效率,后处理简单,具有较好的工业化应用前景。
附图说明
图1为实施例1所得的2-(2-溴-4-氯-苄基)-3,4-二氢-2h-异喹啉-1-酮的核磁共振氢谱。
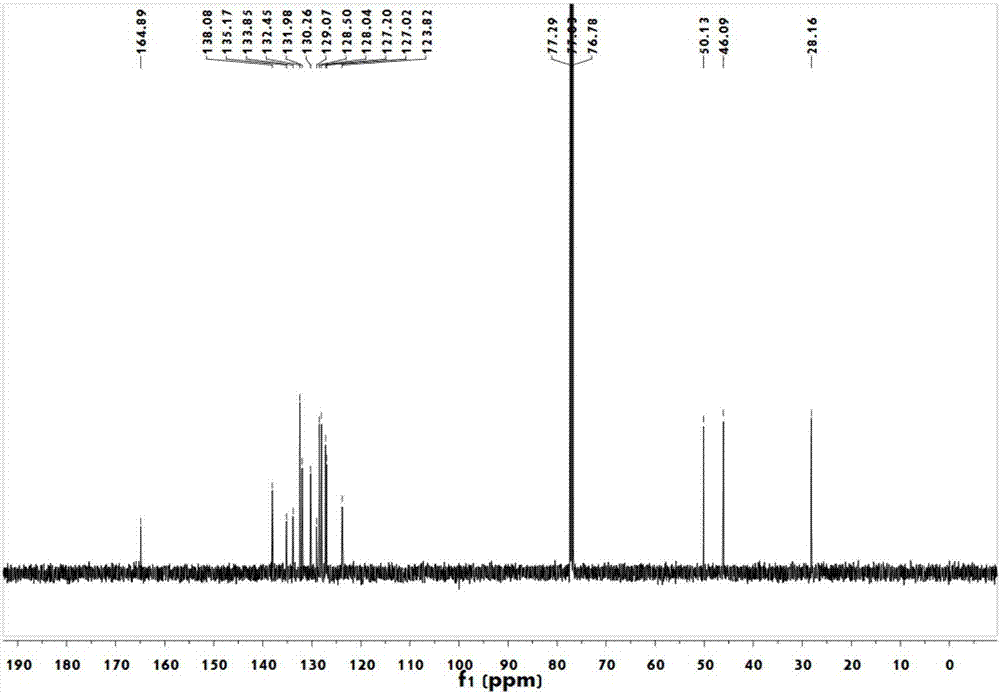
图2为实施例1所得的2-(2-溴-4-氯-苄基)-3,4-二氢-2h-异喹啉-1-酮的核磁共振碳谱。
具体实施方式
下面通过具体实施例并结合附图对本发明进行进一步阐述,但并不限制本发明。
核磁共振氢谱和碳谱测定所用的仪器:核磁共振仪;型号是avanceiii500m,生产厂家是brukercompany。
实施例1
一种2-苄基-3,4-二氢-2h-异喹啉-1-酮衍生物的制备方法,其制备过程具体包括如下步骤:℃
cucl2(16mg)、冰乙酸(12ul)、活化后的分子筛(
采用avanceiii500m核磁共振仪,对上述所得的最终产物进行测定,所得的核磁共振氢谱图、碳谱图分别如图1、图2所示。
采用仪器avanceiii500m核磁共振仪,对上述所得的最终产物进行测定,所得的核磁共振氢谱、碳谱方面的数据如下所示:
1hnmr(500mhz,cdcl3)δ8.13(d,j=7.6hz,1h),7.59(d,j=1.9hz,1h),7.45(t,j=7.4hz,1h),7.37(t,j=7.4hz,1h),7.32(d,j=8.3hz,1h),7.26(dd,j=6.6,1.5hz,1h),7.19(d,j=7.4hz,1h),4.86(s,2h),3.55(t,j=6.6hz,2h),3.00(t,j=6.6hz,2h)ppm.
13cnmr(126mhz,cdcl3)δ=164.89,138.08,135.17,133.85,132.45,131.98,130.26,129.07,128.50,128.04,127.20,127.02,123.82,50.13,46.09,28.16ppm.
从上述所得的核磁共振氢谱、碳谱的数据可以得出最终产物与预测的最终产物相符,氢谱出峰位置和个数全部符合预测情况,碳谱特征峰明显。
实施例2
一种2-苄基-3,4-二氢-2h-异喹啉-1-酮衍生物的制备方法,其制备过程具体包括如下步骤:
cucl2(16mg)、冰乙酸(12ul)、活化后的分子筛(
采用仪器avanceiii500m核磁共振仪,对上述所得的最终产物进行测定,所得的核磁共振氢谱、碳谱方面的数据如下所示:
1hnmr(501mhz,cdcl3)δ8.18(d,j=7.6hz,1h),7.45(t,j=6.9hz,1h),7.39(d,j=7.5hz,1h),7.36(d,j=4.5hz,2h),7.31(s,1h),7.19(d,j=7.4hz,1h),4.83(s,2h),3.51(s,1h),2.96(s,1h)ppm.
13cnmr(126mhz,cdcl3)δ=164.6,138.1,137.5,131.7,129.4,128.6,128.5,128.1,127.4,127.1,126.9,50.5,45.4,28.1ppm.
从上述所得的核磁共振氢谱、碳谱的数据可以得出最终产物与预测的最终产物相符,氢谱出峰位置和个数全部符合预测情况,碳谱特征峰明显。
实施例3
一种2-苄基-3,4-二氢-2h-异喹啉-1-酮衍生物的制备方法,其制备过程具体包括如下步骤:
cucl2(16mg)、冰乙酸(12ul)、活化后的分子筛(
采用仪器avanceiii500m核磁共振仪,对上述所得的最终产物进行测定,所得的核磁共振氢谱、碳谱方面的数据如下所示:
1hnmr(500mhz,cdcl3)δ8.25(d,j=7.5hz,1h),7.54(s,1h),7.48(s,
1h),7.43(s,1h),7.37(s,2h),7.34(s,1h),7.29(d,j=7.4hz,1h),4.88(s,2h),3.61(s,2h),3.08(s,2h)ppm.
13cnmr(126mhz,cdcl3)δ=164.70,139.60,138.02,134.57,131.88,129.97,129.18,128.53,128.05,127.71,127.17,126.98,126.18,50.09,45.59,28.10ppm.
从上述所得的核磁共振氢谱、碳谱的数据可以得出最终产物与预测的最终产物相符,氢谱出峰位置和个数全部符合预测情况,碳谱特征峰明显。
实施例4
一种2-苄基-3,4-二氢-2h-异喹啉-1-酮衍生物的制备方法,其制备过程具体包括如下步骤:
cucl2(16mg)、冰乙酸(12ul)、活化后的分子筛(
采用仪器avanceiii500m核磁共振仪,对上述所得的最终产物进行测定,所得的核磁共振氢谱、碳谱方面的数据如下所示:
1hnmr(500mhz,cdcl3)δ8.25(s,1h),7.52(s,1h),7.46(s,1h),7.33(s,2h),7.25(s,3h),4.87(s,2h),3.58(s,2h),3.03(s,2h),2.45(s,3h)ppm.
13cnmr(126mhz,cdcl3)δ=164.56,138.08,137.13,134.42,131.66,129.48,129.32(s,2c),128.47,128.109(s,2c),127.06,126.88,50.16,45.24,28.12,21.10ppm.
从上述所得的核磁共振氢谱、碳谱的数据可以得出最终产物与预测的最终产物相符,氢谱出峰位置和个数全部符合预测情况,碳谱特征峰明显。
实施例5
一种2-苄基-3,4-二氢-2h-异喹啉-1-酮衍生物的制备方法,其制备过程具体包括如下步骤:
cucl2(16mg)、冰乙酸(12ul)、活化后的分子筛(
采用仪器avanceiii500m核磁共振仪,对上述所得的最终产物进行测定,所得的核磁共振氢谱、碳谱方面的数据如下所示:
1hnmr(500mhz,cdcl3)δ8.12(d,j=7.6hz,1h),7.44(t,j=7.0hz,1h),7.40(d,j=1.8hz,1h),7.38–7.34(m,2h),7.23–7.17(m,2h),4.87(s,2h),3.56(t,j=6.6hz,2h),2.99(t,j=6.6hz,2h)ppm.
13cnmr(126mhz,cdcl3)δ=164.89,138.08,134.23,133.76,133.59,131.96,130.53,129.37,129.09,128.47,127.47,127.19,127.01,47.75,46.15,28.16ppm.
从上述所得的核磁共振氢谱、碳谱的数据可以得出最终产物与预测的最终产物相符,氢谱出峰位置和个数全部符合预测情况,碳谱特征峰明显。
实施例6
一种2-苄基-3,4-二氢-2h-异喹啉-1-酮衍生物的制备方法,其制备过程具体包括如下步骤:
cucl2(16mg)、冰乙酸(12ul)、活化后的分子筛(
采用仪器avanceiii500m核磁共振仪,对上述所得的最终产物进行测定,所得的核磁共振氢谱、碳谱方面的数据如下所示:
1hnmr(500mhz,cdcl3)δ8.17(d,j=7.6hz,0h),7.44(d,j=16.1hz,4h),7.28(d,j=7.6hz,1h),7.20(d,j=7.3hz,1h),7.10(d,j=7.6hz,1h),4.90(s,2h),3.55(s,2h),2.99(s,2h),2.33(s,2h)ppm.
13cnmr(126mhz,cdcl3)δ=164.56,138.08,137.13,134.42,131.66,129.48,129.32(s,2c),128.47,128.10(s,2c),127.06,126.88,50.16,45.24,28.12,21.10ppm.
从上述所得的核磁共振氢谱、碳谱的数据可以得出最终产物与预测的最终产物相符,氢谱出峰位置和个数全部符合预测情况,碳谱特征峰明显。
实施例7
一种2-苄基-3,4-二氢-2h-异喹啉-1-酮衍生物的制备方法,其制备过程具体包括如下步骤:
cucl2(16mg)、冰乙酸(12ul)、活化后的分子筛(
采用仪器avanceiii500m核磁共振仪,对上述所得的最终产物进行测定,所得的核磁共振氢谱、碳谱方面的数据如下所示:
1hnmr(501mhz,cdcl3)δ8.17(d,j=7.6hz,0h),7.44(t,j=7.4hz,1h),7.38(t,j=7.4hz,1h),7.28(d,j=7.8hz,1h),7.19(d,j=7.8hz,2h),4.79(s,2h),3.51(t,j=6.6hz,1h),2.95(t,j=6.6hz,1h),2.66(q,j=7.6hz,1h),1.25(t,j=7.6hz,2h)ppm.
13cnmr(126mhz,cdcl3)δ=164.56,143.51,138.10,134.64,131.68,129.48,128.14(s,5c),127.06,126.99,50.20,45.30,28.54,28.12,15.58ppm.
从上述所得的核磁共振氢谱、碳谱的数据可以得出最终产物与预测的最终产物相符,氢谱出峰位置和个数全部符合预测情况,碳谱特征峰明显。
实施例8
一种2-苄基-3,4-二氢-2h-异喹啉-1-酮衍生物的制备方法,其制备过程具体包括如下步骤:
cucl2(16mg)、冰乙酸(12ul)、活化后的分子筛(
采用仪器avanceiii500m核磁共振仪,对上述所得的最终产物进行测定,所得的核磁共振氢谱、碳谱方面的数据如下所示:
1hnmr(501mhz,cdcl3)δ8.17(d,j=7.2hz,1h),7.60(d,j=7.7hz,1h),7.46(s,1h),7.39(d,j=5.4hz,3h),7.19(d,j=21.1hz,2h),4.94(s,2h),3.58(s,2h),3.02(s,2h)ppm.
13cnmr(126mhz,cdcl3)δ=164.18,164.02,138.15,136.35,132.92,131.87,129.30,128.93,128.50,127.78,127.15,126.98,123.68,50.55,45.92,28.15ppm.
从上述所得的核磁共振氢谱、碳谱的数据可以得出最终产物与预测的最终产物相符,氢谱出峰位置和个数全部符合预测情况,碳谱特征峰明显。
实施例9
一种2-苄基-3,4-二氢-2h-异喹啉-1-酮衍生物的制备方法,其制备过程具体包括如下步骤:
cucl2(16mg)、冰乙酸(12ul)、活化后的分子筛(
采用仪器avanceiii500m核磁共振仪,对上述所得的最终产物进行测定,所得的核磁共振氢谱、碳谱方面的数据如下所示:
1hnmr(500mhz,cdcl3)δ8.26(d,j=7.5hz,1h),7.52(s,1h),7.46(s,1h),7.34(d,j=7.9hz,2h),7.25(d,j=7.8hz,3h),4.87(s,2h),3.58(s,2h),3.03(s,2h)ppm.
13cnmr(126mhz,cdcl3)δ=164.70,139.60,138.0,134.57,131.88,129.97,129.18,128.53,128.05,127.71,127.17,126.98,126.18,50.09,45.59,28.10ppm.
从上述所得的核磁共振氢谱、碳谱的数据可以得出最终产物与预测的最终产物相符,氢谱出峰位置和个数全部符合预测情况,碳谱特征峰明显。
实施例10
一种2-苄基-3,4-二氢-2h-异喹啉-1-酮衍生物的制备方法,其制备过程具体包括如下步骤:
cucl2(16mg)、冰乙酸(12ul)、活化后的分子筛(
采用仪器avanceiii500m核磁共振仪,对上述所得的最终产物进行测定,所得的核磁共振氢谱、碳谱方面的数据如下所示:
1hnmr(501mhz,cdcl3)δ8.19(d,j=7.2hz,1h),7.87(d,j=7.8hz,1h),7.46(s,1h),7.40(d,j=7.3hz,1h),7.33(d,j=4.2hz,2h),7.21(d,j=7.5hz,1h),6.99(d,j=8.0hz,1h),4.88(s,2h),3.54(s,2h),3.01(s,2h)ppm.
13cnmr(126mhz,cdcl3)δ164.78,139.76,139.64,139.29,138.18,131.92,130.10,129.17,128.67,128.54,127.17,127.06,98.98,55.35,45.87,28.17ppm.
从上述所得的核磁共振氢谱、碳谱的数据可以得出最终产物与预测的最终产物相符,氢谱出峰位置和个数全部符合预测情况,碳谱特征峰明显。
实施例11
一种2-苄基-3,4-二氢-2h-异喹啉-1-酮衍生物的制备方法,其制备过程具体包括如下步骤:
cucl2(16mg)、冰乙酸(12ul)、活化后的分子筛(
采用仪器avanceiii500m核磁共振仪,对上述所得的最终产物进行测定,所得的核磁共振氢谱、碳谱方面的数据如下所示:
1hnmr(501mhz,cdcl3)δ8.14(d,j=7.6hz,1h),7.52(d,j=8.7,5.2hz,1h),7.48–7.43(m,1h),7.38(t,j=7.5hz,1h),7.20(d,j=7.4hz,1h),7.11–7.05(m,1h),6.87(td,j=8.4,2.9hz,1h),4.86(s,2h),3.58(t,j=6.6hz,1h),3.03(t,j=6.6hz,1h)ppm.
13cnmr(126mhz,cdcl3)δ164.91,161.37,138.74,138.09,134.10,132.03,129.00,128.55,127.24,127.14,117.39,116.18,116.00,50.79,46.26,28.16ppm.
从上述所得的核磁共振氢谱、碳谱的数据可以得出最终产物与预测的最终产物相符,氢谱出峰位置和个数全部符合预测情况,碳谱特征峰明显。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种回收四甲基氢氧化铵的方法及装置的制造方法
- 四甲基氢氧化铵显影废液再生系统和再生方法
- N-甲基-n-((3r,4r)-4-甲基-1-苄基-3-哌啶基)-7h-吡咯并[2,3-d]嘧啶-4-胺的一种结 ...的制作方法
- 以四乙基氢氧化铵与n,n,n-三甲基金刚烷氢氧化铵混合作为模板剂合成cha的方法
- 1-金刚烷基三甲基氢氧化铵的制备方法
- 一种苄基乙酰氧基化的方法
- 一种四甲基氢氧化铵的提纯回收装置的制造方法
- 一种高纯度四甲基氢氧化铵的提取设备的制造方法
- (2s,5r)-6-苄氧基-7-氧代-1,6-二氮杂-双环[3.2.1]辛烷-2-羧酸钠及其制备方法
- 2-(5-溴-2-甲基苄基)-5-(4-氟苯基)噻吩的制备方法