反应性液晶原、液晶组合物及包含它的液晶显示装置的制作方法
本发明涉及一种反应性液晶原、液晶组合物及包含它的液晶显示装置。
背景技术:
液晶显示装置具有两个透明电极基板之间注入液晶的结构,通过基于电场使液晶取向的方式来驱动。这种液晶显示装置在目前开发出的各种平板显示器中市占率最高。
驱动液晶显示装置的方式大致分为超扭曲向列(tn,twistednematic)方式、垂直取向(va,verticalalignment)方式及平面转换(ips,in-plane-switching)方式,根据tn、va、ips模式液晶分子的配置方法和基于电压的液晶分子运动发生改变。
va模式液晶显示装置在两个透明电极基板之间包含具有负介电常数各向异性的液晶分子。所述液晶分子在未施加电压的状态下相对于电极表面垂直取向,而在施加电压的状态下相对于电极表面平行取向。因此,va模式液晶显示装置根据基于施加电压与否的液晶分子的取向,可以控制来自背光的光的开与关。但是,对于va模式液晶显示装置,如果不预先确定电压施加于电极时平行取向的方向性,就会存在响应速度非常慢的缺陷。
为了解决这种问题推出了聚合物稳定取向(psa,polymerstabilizedaligned)或聚合物稳定液晶垂直取向(ps-va,polymerstabilized-verticalaligned)模式液晶显示装置。对于psa或ps-va模式液晶显示装置,将主体液晶(hostliquidcrystal)和可紫外线聚合的液晶原化合物(reactivemesogen)混合后注入液晶单元,对液晶单元施加电压使主体液晶和可聚合的液晶原化合物取向后,通过照射光线使可聚合的液晶原化合物聚合,从而在未施加电压的状态下也会使主体液晶保持一定倾斜,当再施加电压时,主体液晶朝倾斜方向快速取向,从而可以实现快速响应。用于psa或ps-va模式液晶显示装置的反应性液晶原要求相对于主体液晶显示出高溶解度,并显示出高光反应效率。
技术实现要素:
技术问题
本发明旨在提供一种可显示出优异的曝光效率的反应性液晶原。
本发明还提供包含所述反应性液晶原的液晶组合物及包含它的液晶显示装置。
解决问题的方法
下面对根据本发明的具体实施方案的反应性液晶原及其用途等进行说明。
根据本发明的一个实施方案提供一种由下述化学式1表示的反应性液晶原。
[化学式1]
在所述化学式1中,
x1和x2各自独立地为氢或卤素,
y为-o-、-s-、-nh-或-sih2-,
l为0或1的整数,
p1和p2为彼此相同或不同的聚合性官能团,
sp1和sp2各自独立地为单键或选自具有1至15个碳原子的直链亚烷基中的任意一个二价自由基、或者所述二价自由基中至少一个氢被卤素或氰基取代或至少一个-ch2-以氧原子不直接连接的方式被-c≡c-、-ch=ch-、-o-、-co-o-、-o-co-或-o-co-o-替代的二价自由基。
所述反应性液晶原与主体液晶混合、取向及聚合而形成预倾角,从而可以提供psa或ps-va模式液晶显示装置,并且由于以取向的状态聚合,可以提供各种光学膜。特别是,所述反应性液晶原显示出优异的机械和热稳定性,从而可以提供寿命及可靠性得到提高的液晶显示装置和机械和热物性优异的各种光学膜。
此外,所述反应性液晶原显示出较高的紫外线吸光度,因此通过较少的曝光量快速聚合,从而可以节省液晶显示装置或光学膜的制备时间及费用。另外,包含含有所述反应性液晶原的液晶组合物的液晶显示装置可以显示出优异的响应速度和高电压保持率。
在所述化学式1中,x1和x2各自独立地为氢或–f、-cl、-br或–i等卤素,更具体地x1和x2各自独立地可为氢或-f。
在所述化学式1中,p1和p2各自独立地可为(甲基)丙烯酰基、(甲基)丙烯酰氧基、乙烯基、乙烯氧基或环氧基。
在所述化学式1中,sp1和sp2各自独立地可为单键、-(ch2)a-、-o-(ch2)b-、-(ch2)b-o-、-o-(ch2)d-o-或-[o-(ch2)e]h-o-,其中a为1至15的整数,b各自独立地为1至14的整数,d为1至13的整数,e为1至4的整数,h为1至13的整数。具体地,sp1和sp2各自独立地可为单键、亚甲基、亚乙基或丙烯基,更具体地可为单键或亚甲基。
另外,在所述化学式1中,y可为-o-或-s-。
作为一个实例,所述由化学式1表示的反应性液晶原可为由下述化学式2或3表示的化合物。
[化学式2]
[化学式3]
在所述化学式2及3中,
x1和x2各自独立地为氢或-f,
y为-o-或-s-,
n和m各自独立地为0、1或2的整数,
r1和r2各自独立地为氢或甲基。
在更具体的实例中,所述由化学式1表示的反应性液晶原可为选自由下述化学式1-1至1-64组成的组中的化合物。
另外,根据本发明的另一个实施方案提供包含至少一种所述反应性液晶原的液晶组合物。
相对于100重量份的总液晶组合物,所述液晶组合物可包含0.01至10重量份或0.05至5重量份或0.07至3重量份或0.1至1重量份的所述反应性液晶原。
如果反应性液晶原的含量少于所述范围,则与主体液晶混合及聚合后,无法形成适当的预倾角,如果多于所述范围,则曝光后仍残留未反应的反应性液晶原,可能会影响主体液晶的物性或者降低液晶显示装置或光学膜的可靠性。
所述液晶组合物还可包含由下述化学式4表示的液晶化合物,当然也可包含多种用化学式4定义的液晶化合物。
[化学式4]
在所述化学式4中,r21和r22各自独立地为氢及具有1至15个碳原子的烷基中的任意一个自由基、或者所述自由基中的至少一个氢被卤素取代或至少一个-ch2-以氧原子不直接连接的方式被-c≡c-、-ch=ch-、-o-、-co-o-、-o-co-或-o-co-o-替代的自由基,
a3和a4环各自独立地为1,4-亚环己基、亚四氢吡喃基(tetrahydropyranylene)或1,4-亚苯基。
在没有特别限制的情况下,本说明书中下列术语定义如下。
卤素(halogen)可以是氟(f)、氯(cl)、溴(br)或碘(i)。
具有1至15个碳原子的烷基自由基可以是直链、支链或环状烷基自由基。具体地,具有1至15个碳原子的烷基自由基可以是具有1至10个碳原子的直链烷基自由基;具有1至5个碳原子的直链烷基自由基;具有3至10个碳原子的支链或环状烷基自由基;或者具有3至5个碳原子的支链或环状烷基自由基。更具体地,具有1至15个碳原子的烷基自由基可以是甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、正戊基、异戊基或环己基等。
此外,具有1至15个碳原子的烷基自由基可被所述自由基的至少一个-ch2-被-c≡c-、-ch=ch-、-o-、-co-o-、-o-co-或–o-co-o-取代的自由基替代。作为一个实例,甲基自由基可被甲基自由基(-ch2-h)的-ch2-被-ch=ch-取代的乙烯基自由基(-ch=ch-h)替代。但是,所述自由基中至少一个-ch2-可以以氧原子不直接连接的方式被所述的取代基取代。
另外,具有1至15个碳原子的烷基自由基可被所述自由基的至少一个h被卤素取代的自由基替代。作为一个实例,甲基自由基可被甲基自由基(-ch3)的所有h被f取代的全氟甲基自由基(-cf3)替代。
作为所述化学式4的液晶化合物可以使用选自下述结构的液晶化合物中的化合物,以保持较高的电阻率,并且可以有利地控制透明点、旋转粘度、折射率各向异性、介电常数各向异性。
在所述式中,r21和r22与化学式4中所定义的相同。
为了实现上述的物性,相对于100重量份的总液晶组合物,可包含10至50重量份、20至40重量份或25至35重量份的所述化学式4的化合物。
另外,所述液晶组合物还可包含由下述化学式5表示的液晶化合物,当然也可包含多种用化学式5定义的液晶化合物。
[化学式5]
在所述化学式5中,r31和r32各自独立地为氢及具有1至15个碳原子的烷基中的任意一个自由基、或者所述自由基中的至少一个氢被卤素取代或至少一个-ch2-以氧原子不直接连接的方式被-c≡c-、-ch=ch-、-o-、-co-o-、-o-co-或-o-co-o-替代的自由基,
a5和a7环各自独立地为1,4-亚环己基、亚四氢吡喃基或1,4-亚苯基,
a6环为1,4-亚环己基、亚四氢吡喃基、1,4-亚苯基或至少一个h被卤素取代的1,4-亚苯基,
o为1或2的整数。
在所述化学式5中,当o为2时,两个a6环可以彼此相同或不同。
作为这种化学式5的液晶化合物可以使用选自下述结构的液晶化合物中的化合物,以保持较高的电阻率,并且可以有利地控制透明点、旋转粘度、折射率各向异性、介电常数各向异性。
在所述式中,r31和r32与化学式5中所定义的相同。
另外,在本说明书的化学式中括号“()”表示可被括号内的取代基取代。更具体地,-(f)表示在其部位上可以结合氢或氟。
为了实现上述的物性,相对于100重量份的总液晶组合物,可包含5至40重量份、5至20重量份或5至10重量份的所述化学式5的液晶化合物。
另外,所述液晶组合物还可包含由下述化学式6表示的液晶化合物,当然也可包含多种用化学式6定义的液晶化合物。
[化学式6]
在所述化学式6中,r41和r42各自独立地为氢及具有1至15个碳原子的烷基中的任意一个自由基、或者所述自由基中的至少一个氢被卤素取代或至少一个-ch2-以氧原子不直接连接的方式被-c≡c-、-ch=ch-、-o-、-co-o-、-o-co-或-o-co-o-替代的自由基,
a8和a9环各自独立地为1,4-亚环己基、亚四氢吡喃基、1,4-亚苯基或至少一个h被卤素取代的1,4-亚苯基,
m为0、1或2的整数。
作为这种化学式6的液晶化合物可以使用选自下述结构的液晶化合物中的化合物,以保持较高的电阻率,并且可以有利地控制透明点、折射率各向异性、介电常数各向异性。
在所述式中,r41和r42与化学式6中所定义的相同。
为了实现上述的物性,相对于100重量份的总液晶组合物,可包含10至75重量份、30至70重量份或45至65重量份的所述化学式6的液晶化合物。
另外,所述液晶组合物还可包含由下述化学式7表示的液晶化合物,当然也可包含多种用化学式7定义的液晶化合物。
[化学式7]
在所述化学式7中,r51和r52各自独立地为氢及具有1至15个碳原子的烷基中的任意一个自由基、或者所述自由基中的至少一个氢被卤素取代或至少一个-ch2-以氧原子不直接连接的方式被-c≡c-、-ch=ch-、-o-、-co-o-、-o-co-或-o-co-o-替代的自由基,
a10、a11和a12环各自独立地为1,4-亚环己基、亚四氢吡喃基、1,4-亚苯基或至少一个h被卤素取代的1,4-亚苯基,
z1和z2各自独立地为-ch2ch2-、-ch=ch-、-ch2o-、-och2-、-c≡c-、-ch2cf2-、-chfchf-、-cf2ch2-、-ch2chf-、-chfch2-、-c2f4-、-o-、-coo-、-oco-、-cf2o-或-ocf2-,
n1和n3各自独立地为0或1的整数,n1与n3之和为1或2,
n2和n4各自独立地为0、1或2的整数。
作为这种化学式7的液晶化合物可以使用选自下述结构的液晶化合物中的化合物,以保持较高的电阻率,并且可以有利地控制透明点、旋转粘度、折射率各向异性、介电常数各向异性。
在所述式中,r51和r52与化学式7中所定义的相同。
特别是,作为所述化学式7的液晶化合物使用由所述7-1表示的液晶化合物时,可显示出更低的旋转粘度,因此非常有利于提供快速响应用液晶组合物。
为了实现上述的物性,相对于100重量份的总液晶组合物,可包含0至30重量份、0至20重量份或0至15重量份的所述化学式7的化合物。
除了液晶化合物之外,所述液晶组合物还可包含本发明所属技术领域中常用的各种添加剂。这种添加剂例如有抗氧化剂或紫外线吸收剂等。
另外,根据本发明的另一个实施方案提供一种包含所述液晶组合物的液晶显示装置。所述液晶显示装置在所述反应性液晶原与主体液晶混合后取向的状态下聚合而形成预倾角,从而能够以psa或ps-va模式驱动。
特别是,所述反应性液晶原显示出优异的机械和热稳定性,从而可以提高液晶显示装置的寿命及可靠性,而且显示出较高的紫外线吸光度,因此通过较少的曝光量快速聚合,从而可以节省液晶显示装置的制备时间及费用。另外,包含含有所述反应性液晶原的液晶组合物的液晶显示装置可以显示出优异的响应速度和高电压保持率
所述反应性液晶原除了用于液晶显示装置,还可用于形成光学膜,可以显示出优异的机械和热物性,预计将用于各种领域。
发明效果
根据本发明的反应性液晶原与主体液晶混合、取向及聚合而形成预倾角,从而可以提供psa或ps-va模式液晶显示装置,并且由于以取向的状态聚合,可以提供各种光学膜。特别是,所述反应性液晶原显示出优异的机械和热稳定性,从而可以提供寿命及可靠性得到提高的液晶显示装置及各种光学膜。
此外,所述反应性液晶原显示出较高的紫外线吸光度,因此通过较少的曝光量快速聚合,从而可以节省液晶显示装置或光学膜的制备时间及费用。另外,包含含有所述反应性液晶原的液晶组合物的液晶显示装置可以显示出优异的响应速度和高电压保持率。
附图说明
图1至图6分别是合成例1至6中合成的rm-1至rm-6的nmr光谱。
图7是根据试验例1测定的反应性液晶原的uv吸收光谱。
图8是示出根据实施例1至3及比较例1制备的va-测试单元基于曝光时间(min)的预倾角(°)变化的曲线图。
图9是示出根据实施例4至6及比较例2制备的va-测试单元基于曝光时间(min)的预倾角(°)变化的曲线图。
具体实施方式
下面通过本发明的具体实施例更具体地描述本发明的作用及效果。但,下述实施例是本发明的示例而已,本发明的权利范围不限于下述实施例。
合成例1:反应性液晶原的制备(制备rm-1)
1)化合物3的合成(1-溴-4-(2,2-二甲氧基乙氧基)苯)
将4-溴苯酚1(4-bromophenol,30.0g,173.4mmol)、koh(11.7g,208.1mmol)、dmso(90.0ml,3ml/g)进行混合后,再加入溴代乙醛缩二甲醇2(bromoacetaldehydedimethylacetal,32.2g,190.7mmol),回流反应1小时。反应结束后,冷却至常温,再加入水。用乙酸乙酯(etoac)提取有机层后,对有机层用无水硫酸钠(anhydrousna2so4)进行干燥及过滤并减压浓缩。将浓缩残渣用硅胶柱层析法(己烷:etoac=10:1)进行纯化得到无色液体化合物3(38.9g,收率86%)。
1h-nmr(cdcl3,varian400mhz):δ7.36(2h,d,j=9.2hz),6.80(2h,d,j=8.8hz),4.70(1h,t,j=5.2hz),3.96(2h,d,j=5.2hz),3.45(6h,s)
2)化合物4的合成(5-溴苯并呋喃)
甲苯(390.0ml,10ml/g)中加入多聚磷酸(polyphosphoricacid,38.9g,1g/g),再加入化合物3(1-溴-4-(2,2-二甲氧基乙氧基)苯,38.9g,149.0mmol),然后在80℃下搅拌2小时。反应结束后,反应物中加入2n的naoh水溶液进行中和,然后分离出有机层。对有机层用无水硫酸钠进行干燥及过滤并减压浓缩。将浓缩残渣用硅胶柱层析法(己烷)进行纯化得到化合物4(18.3g,收率62%)。
1h-nmr(cdcl3,varian400mhz):δ7.72(1h,d,j=2.4hz),7.61(1h,d,j=1.6hz),7.37(2h,s),6.71(1h,d,j=2.0hz)
3)化合物6的合成(4-(苯并呋喃-5-基)苯甲酸甲酯)
化合物4(5-溴苯并呋喃,8.5g,43.4mmol)、化合物5(4-(甲氧基羰基)苯基硼酸,8.6g,47.8mmol)溶解于meoh(9.0ml,1ml/g)、甲苯(90.0ml,10ml/g)中,再加入cs2co3(28.1g,86.3mmol),然后充入氮气。在氮气下加入pd(pph3)4(2.5g,2.2mmol),然后回流搅拌2小时。反应结束后,将反应物利用硅藻土过滤后进行浓缩,然后通过再结晶得到白色固体化合物6(9.7g,收率89%)。
1h-nmr(cdcl3,varian400mhz):δ8.11(1h,d,j=8.8hz),7.83(1h,d,j=1.2hz),7.68-7.70(3h,m),7.54-7.60(2h,m),6.83(1h,dd,j=2.0,0.8hz),3.95(3h,s)
4)化合物7的合成((4-(苯并呋喃-5-基)苯基)甲醇)
将化合物6(4-(苯并呋喃-5-基)苯甲酸甲酯,9.7g,38.5mmol)溶解于thf(200.0ml,20ml/g)中,然后在0℃下一点一点加入氢化铝锂(lithiumaluminumhydride,1.6g,42.3mmol)。加入完毕后,在常温下反应2小时。反应结束后,反应物中加入水进行费希尔后处理(fisherwork-up),然后对得到有机层通过加入无水硫酸镁(anhydrousmgso4)进行干燥、过滤及减压浓缩,从而得到白色固体化合物7(8.6g,定量)。
1h-nmr(cdcl3,varian400mhz):δ7.79(1h,d,j=1.2hz),7.66(1h,d,j=2.4hz),7.61-7.63(2h,m),7.50-7.57(2h,m),7.45(2h,d,j=8.4hz),6.82(1h,dd,j=2.4,0.8hz),4.75(2h,d,j=6.0hz),1.65(1h,t,j=6.0hz)
5)化合物8的合成(5-(4-((四氢-2h-吡喃-2-基氧基)甲基)苯基)苯并呋喃)
将化合物7((4-(苯并呋喃-5-基)苯基)甲醇,8.6g,38.4mmol)溶解于thf(170ml,20ml/g)中,再将对甲苯磺酸(p-toluenesulfonicacidmonohydrate,0.360g,1.9mmol)、3,4-二氢-2h-吡喃(3,4-dihydro-2h-pyran,7.0ml,76.7mmol)按顺序加入,然后在常温下反应2.5小时。反应物中加入饱和氯化铵溶液结束反应。利用乙酸乙酯提取有机层,对有机层用无水硫酸钠进行干燥及过滤并减压浓缩。对浓缩残渣利用硅胶柱层析法(己烷:etoac=10:1)得到无色液体化合物8(9.4g,收率80%)。
1h-nmr(cdcl3,varian400mhz):δ7.78(1h,s),7.65-7.66(1h,m),7.51-7.62(4h,m),7.45(2h,d,j=8.4hz),6.82(1h,d,j=1.6hz),4.84-4.87(1h,m),4.76-4.77(1h,m),4.55-4.58(1h,m),3.94-3.99(1h,m),3.56-3.60(1h,m),1.55-1.88(6h,m)
6)化合物9的合成((5-(4-((四氢-2h-吡喃-2-基氧基)甲基)苯基)苯并呋喃-2-基)甲醇)
将化合物8(5-(4-((四氢-2h-吡喃-2-基氧基)甲基)苯基)苯并呋喃,11.8g,38.3mmol)溶解于thf(240.0ml,20ml/g)后冷却至-78℃,其中缓慢滴入正丁基锂(36.0ml,57.6mmol,1.6m的己烷溶液),并在-78℃下搅拌30分钟。所述混合溶液中加入多聚甲醛(p-formaldehyde,8.5g,0.283mol)。然后,将得到的反应物升温至常温,并进一步搅拌3小时。反应物中加入饱和氯化铵溶液结束反应。利用有机溶剂分离出有机层,对有机层用无水硫酸钠进行干燥及过滤并减压浓缩。将浓缩残渣用硅胶柱层析法(己烷:etoac=4:1)进行纯化得到白色固体化合物9(7.2g,收率56%)。
1h-nmr(cdcl3,varian400mhz):δ7.74(1h,s),7.59(2h,d,j=8.0hz),7.51(2h,s),7.44(2h,d,j=7.6hz),6.71(1h,s),4.84(1h,d,j=12.4hz),4.79-4.79-4.80(2h,m),4.74-4.76(1h,m),4.55(1h,d,j=12.0hz),3.93-3.98(1h,m),3.55-3.59(1h,m),1.55-1.93(7h,m)
7)化合物10的合成((4-(2-(羟甲基)苯并呋喃-5-基)苯基)甲醇)
将化合物9((5-(4-((四氢-2h-吡喃-2-基氧基)甲基)苯基)苯并呋喃-2-基)甲醇,7.2g,21.3mmol)溶解于meoh(70.0ml,10ml/g)后,再加入对甲苯磺酸(p-toluenesulfonicacidmonohydrate,0.400g,2.1mmol),在常温下搅拌2小时。反应结束后,反应物中加入水,然后分离出有机层。对得到的有机层用无水硫酸钠进行干燥及过滤并减压浓缩。将浓缩残渣用硅胶柱层析法(mc:meoh=20:1)进行纯化得到白色固体化合物10(4.8g,收率99%)。
1h-nmr(cdcl3,varian400mhz):δ7.74(1h,s),7.61(2h,d,j=8.4hz),7.45(2h,d,j=8.0hz),7.52(2h,s),7.44-7.46(2h,m),6.72(1h,s),4.80(1h,d,j=6.4hz),4.75(1h,d,j=6.0hz),1.90(1h,t,j=6.4hz),1.66(1h,t,j=6.0hz)
8)化合物rm-1的合成(4-(2-(甲基丙烯酰氧基甲基)苯并呋喃-5-基)甲基丙烯酸苄基酯)
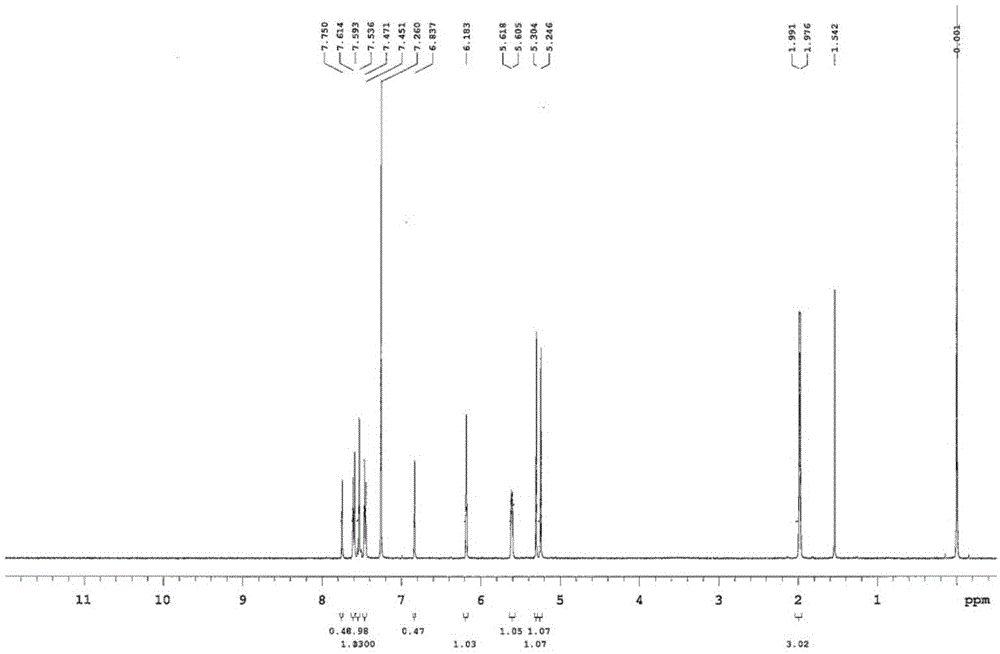
将化合物10((4-(2-(羟甲基)苯并呋喃-5-基)苯基)甲醇,3.3g,14.5mmol)溶解于无水thf(100ml,30ml/g)后,在0℃下将dmap(dimethylaminopyridine,0.200g,1.5mmol)、tea(triethylamine,7.1ml,50.9mmol)、甲基丙烯酰氯(methacryloylchloride,4.3ml,43.6mmol)按顺序加入,再升温至常温反应3小时。反应结束后,反应物中加入饱和碳酸氢钠溶液,然后分离出有机层。对有机层用无水硫酸钠进行干燥及过滤并减压浓缩。将浓缩残渣用硅胶柱层析法(己烷:etoac=8:1)进行纯化得到白色固体化合物rm-1(1.1g,收率20%)。rm-1的nmr光谱示于图1中。
1h-nmr(cdcl3,varian400mhz):δ7.75(1h,s),7.60(2h,d,j=8.0hz),7.55(2h,s),7.46(2h,d,j=8.4hz),6.83(1h,s),6.18(2h,s),5.61-5.62(2h,m),5.30(2h,s),5.24(2h,s),1.99(3h,s),1.96(3h,s)
合成例2:反应性液晶原的制备(制备rm-2)
1)化合物11的合成(2-(4-溴苯氧基)四氢-2h-吡喃)
将4-溴苯酚1(10.0g,57.8mmol)溶解于thf(100.0ml,10ml/g)后,将对甲苯磺酸(p-toluenesulfonicacidmonohydrate,0.110g,0.6mmol)、3,4-二氢-2h-吡喃(3,4-dihydro-2h-pyran,10.5ml,0.115mol)按顺序加入,然后在常温下反应10小时。反应物中加入饱和氯化铵溶液结束反应。利用乙酸乙酯提取有机层,对有机层用无水硫酸钠进行干燥及过滤并减压浓缩。将浓缩残渣用硅胶柱层析法(己烷:etoac=10:1)进行纯化得到无色液体化合物11(10.7g,收率72%)。
1h-nmr(cdcl3,varian400mhz):δ7.37(2h,d,j=8.8hz),6.94(2h,d,j=8.8hz),5.36-5.38(1h,m),3.84-3.90(1h,m),3.57-3.62(1h,m),1.94-2.03(1h,m),1.84-1.86(1h,m),1.57-1.74(4h,m)
2)化合物13的合成(4,4,5,5-四甲基-2-(4-(四氢-2h-吡喃-2-基氧基)苯基)-1,3,2-二氧硼戊环)
将化合物11(2-(4-溴苯氧基)四氢-2h-吡喃,10.7g,41.6mmol)溶解于thf(220.0ml,20ml/g)后冷却至-78℃,其中缓慢滴入正丁基锂(21.6ml,54.1mmol,2.5m的己烷溶液),并在-78℃下搅拌30分钟。所述混合溶液中缓慢滴入2-异丙氧基-4,4,5,5-四甲基-1,3,2-二氧硼戊环12(2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane,11.0ml,54.1mmol)。然后,将得到的反应物从-78℃升温至常温,并进一步搅拌2.5小时。反应物中加入饱和氯化铵溶液结束反应。利用有机溶剂分离出有机层,对有机层用无水硫酸钠进行干燥及过滤并减压浓缩。将浓缩残渣用硅胶柱层析法(己烷:etoac=10:1)进行纯化得到白色固体化合物13(9.4g,收率74%)。
1h-nmr(cdcl3,varian400mhz):δ7.74(2h,d,j=8.8hz),7.04(2h,d,j=8.4hz),5.48-5.49(1h,m),3.85-3.91(1h,m),3.57-3.61(1h,m)1.96-2.05(1h,m),1.84-1.88(2h,m),1.58-1.71(3h,m),1.33(12h,s)
3)化合物14的合成(5-(4-(四氢-2h-吡喃-2-基氧基)苯基)苯并呋喃)
将化合物4(5-溴苯并呋喃,5.4g,27.5mmol)、化合物13(4,4,5,5-四甲基-2-(4-(四氢-2h-吡喃-2-基氧基)苯基)-1,3,2-二氧硼戊环,7.6g,25.0mmol)溶解于meoh(8.0ml,1ml/g)、甲苯(80.0ml,10ml/g)中,再加入cs2co3(16.3g,50.0mmol),然后充入氮气。在氮气下加入pd(pph3)4(1.4g,1.2mmol),然后回流搅拌3小时。反应结束后,将反应物利用硅藻土过滤后进行减压浓缩,再用硅胶柱层析法(己烷:etoac=5:1)进行纯化得到白色固体化合物14(5.3g,收率72%)。
1h-nmr(cdcl3,varian400mhz):δ7.74(1h,s),7.62(1h,d,j=2.0hz),7.47-7.54(4h,m),7.13(2h,d,j=8.4hz),6.81(1h,s),5.47-5.48(1h,m),3.95(1h,td,j=2.8,12.8hz),3.62-3.65(1h,m),2.01-2.06(1h,m),1.88-1.90(2h,m),1.61-1.70(3h,m)
4)化合物15的合成((5-(4-(四氢-2h-吡喃-2-基氧基)苯基)苯并呋喃-2-基)甲醇)
将化合物14(5-(4-(四氢-2h-吡喃-2-基氧基)苯基)苯并呋喃,3.6g,12.2mmol)溶解于thf(72.0ml,20ml/g)后冷却至-78℃,其中缓慢滴入正丁基锂(7.3ml,18.3mmol,2.5m的己烷溶液),并在-78℃下搅拌30分钟。所述混合溶液中加入多聚甲醛(p-formaldehyde,2.7g,54.1mmol)。然后,将得到的反应物升温至常温,并进一步搅拌5小时。反应物中加入饱和氯化铵溶液结束反应。利用有机溶剂分离出有机层,对有机层用无水硫酸钠进行干燥及过滤并减压浓缩。将浓缩残渣用硅胶柱层析法(己烷:etoac=3:1)进行纯化得到白色固体化合物15(2.1g,收率53%)。
1h-nmr(cdcl3,varian400mhz):δ7.68-7.69(1h,m),7.45-7.53(4h,m),7.13(2h,d,j=8.4hz),6.69(1h,s),5.47-5.48(1h,m),4.79(2h,d,j=5.6hz),3.95(1h,td,j=2.8,12.4hz),3.62-3.66(1h,m),1.88-2.06(4h,m),1.59-1.76(3h,m)
5)化合物16的合成(4-(2-(羟甲基)苯并呋喃-5-基)苯酚)
将化合物15((5-(4-(四氢-2h-吡喃-2-基氧基)苯基)苯并呋喃-2-基)甲醇,2.1g,6.5mmol)溶解于meoh(42.0ml,20ml/g)后,再加入对甲苯磺酸(p-toluenesulfonicacidmonohydrate,0.123g,0.6mmol),在常温下搅拌1.5小时。反应结束后,反应物中加入水,然后分离出有机层。对得到的有机层用无水硫酸钠进行干燥及过滤并减压浓缩。对浓缩残渣进行再结晶得到白色固体化合物16(1.4g,收率88%)。
1h-nmr(cdcl3,varian400mhz):δ7.68(1h,s),7.44-7.50(4h,m),6.91(2h,d,j=8.8hz),6.70(1h,s),4.80(3h,d,j=6.0hz),4.73(1h,s)
6)化合物rm-2的合成((5-(4-(甲基丙烯酰氧基)苯基)苯并呋喃-2-基)甲基丙烯酸甲酯)
将化合物16(4-(2-(羟甲基)苯并呋喃-5-基)苯酚,1.4g,5.8mmol)溶解于无水thf(42.0ml,30ml/g)后,在0℃下将dmap(0.071g,0.6mmol)、tea(2.8ml,20.4mmol)、甲基丙烯酰氯(methacryloylchloride,1.7ml,17.5mmol)按顺序加入,再升温至常温反应2小时。反应结束后,反应物中加入饱和碳酸氢钠溶液,然后分离出有机层。对有机层用无水硫酸钠进行干燥及过滤并浓缩,再用硅胶柱层析法(己烷:etoac=10:1)进行纯化得到白色固体化合物rm-2(0.9g,收率41%)。rm-2的nmr光谱示于图2中。
1h-nmr(cdcl3,varian400mhz):δ7.74(1h,s),7.61(2h,d,j=8.8hz),7.54(2h,q,j=8.4hz),7.20(2h,d,j=8.8hz),6.84(1h,s),6.38(1h,s),6.18(1h,s),5.78(1h,s),5.62(1h,s),5.30(2h,s),2.09(3h,s),1.98(3h,s)
合成例3:反应性液晶原的制备(制备rm-3)
1)化合物18的合成(1-溴-4-(2,2-二甲氧基乙氧基)-2,3-二氟苯)
将4-溴-2,3-二氟苯酚17(4-bromo-2,3-diflurophenol,100.0g,478.5mmol)、koh(32.2g,574.2mmol)、dmso(300.0ml,3ml/g)进行混合,然后加入溴代乙醛缩二甲醇2(bromoacetaldehydedimethylacetal,89.0g,526.3mmol),回流反应1小时。反应结束后,冷却至常温,并加入饱和氯化铵溶液。用有机溶剂提取有机层后,对有机层用无水硫酸钠进行干燥及过滤并减压浓缩。将浓缩残渣用硅胶柱层析法(己烷:etoac=10:1)进行纯化得到无色液体化合物18(90.4g,收率64%)。
1h-nmr(cdcl3,varian400mhz):δ7.20(1h,td,j=2.4,9.2hz),6.69(1h,td,j=2.4,9.2hz),4.71(1h,t,j=5.2hz),4.05(2h,d,j=5.6hz),3.47(6h,s)
2)化合物19的合成(5-溴-6,7-二氟苯并呋喃)
多聚磷酸(polyphosphoricacid,50.0g,1g/g)和氯苯(500.0ml,10ml/g)的混合溶液中加入化合物18(50.0g,168.3mmol)后,回流反应3小时。反应结束后,将反应物利用硅藻土过滤并加入饱和碳酸氢钠溶液,然后分离出有机层。对得到的有机层用无水硫酸钠进行干燥及过滤并减压浓缩。将浓缩残渣用硅胶柱层析法(己烷)进行纯化得到固体化合物19(8.3g,收率21%)。
1h-nmr(cdcl3,varian400mhz):δ7.68(1h,d,j=2.0hz),7.54(1h,dd,j=2.0,5.6hz),6.75(1h,t,j=2.0hz)
3)化合物20的合成(6,7-二氟-5-(4-(四氢-2h-吡喃-2-基氧基)苯基)苯并呋喃)
将化合物19(5-溴-6,7-二氟苯并呋喃,8.3g,35.6mmol)、化合物13(4,4,5,5-四甲基-2-(4-(四氢-2h-吡喃-2-基氧基)苯基)-1,3,2-二氧硼戊环,9.9g,32.4mmol)溶解于甲苯(99.0ml,10ml/g)中,再加入cs2co3(21.1g,64.8mmol),然后充入氮气。在氮气下加入pd(pph3)4(1.9g,1.6mmol)后,回流搅拌17小时。反应结束后,将反应物利用硅藻土过滤并进行浓缩,再用硅胶柱层析法(己烷:etoac=20:1)进行纯化得到黄色液体化合物20(8.5g,收率79%)。
1h-nmr(cdcl3,varian400mhz):δ7.66(1h,d,j=2.0hz),7.46(2h,d,j=8.8hz),7.33(1h,d,j=1.6hz),7.15(2h,d,j=8.8hz),6.79(1h,d,j=2.0hz),5.49(1h,d,j=2.8hz),3.94(1h,td,j=2.8,8.2hz),3.62-3.67(1h,m),1.99-2.08(1h,m),1.88-1.92(2h,m),1.61-1.76(3h,m)
4)化合物21的合成((6,7-二氟-5-(4-(四氢-2h-吡喃-2-基氧基)苯基)苯并呋喃-2-基)甲醇)
将化合物20(6,7-二氟-5-(4-(四氢-2h-吡喃-2-基氧基)苯基)苯并呋喃,8.5g,25.7mmol)溶解于thf(170.0ml,20ml/g)后冷却至-78℃,其中缓慢滴入正丁基锂(15.4ml,38.6mmol,2.5m的己烷溶液),并在-78℃下搅拌30分钟。所述混合溶液中加入多聚甲醛(p-formaldehyde,5.7g,190.4mmol)。然后,将得到的反应物升温至常温,并进一步搅拌12小时。反应物中加入饱和氯化铵溶液结束反应。利用有机溶剂分离出有机层,对有机层用无水硫酸钠进行干燥及过滤并减压浓缩。将浓缩残渣用硅胶柱层析法(己烷:etoac=10:1)进行纯化得到黄色液体化合物21(5.2g,收率56%)。
1h-nmr(cdcl3,varian400mhz):δ7.44(2h,d,j=7.2hz),7.26-7.27(1h,m),7.13(2h,d,j=8.4hz),6.67(1h,d,j=2.8hz),5.48(1h,t,j=2.8hz),4.78(2h,d,j=4.8hz),3.94(1h,td,j=2.8,8.8hz),3.62-3.66(1h,m),2.19-2.24(1h,m),2.01-2.04(2h,m),1.87-1.91(1h,m),1.60-1.73(3h,m)
5)化合物22的合成(4-(6,7-二氟-2-(羟甲基)苯并呋喃-5-基)苯酚)
将化合物21((6,7-二氟-5-(4-(四氢-2h-吡喃-2-基氧基)苯基)苯并呋喃-2-基)甲醇,5.2g,14.4mmol)溶解于thf(104.0ml,20ml/g)后,再加入对甲苯磺酸(p-toluenesulfonicacidmonohydrate,0.274g,1.4mmol),在常温下搅拌1小时。反应结束后,反应物中加入水,然后分离出有机层。对得到的有机层用无水硫酸钠进行干燥及过滤并减压浓缩。接着,对浓缩残渣进行再结晶得到黄色液体化合物22(3.2g,收率80%)。
1h-nmr(cdcl3,varian400mhz):δ7.42(2h,d,j=7.2hz),7.25-7.26(1h,m),6.93(2h,d,j=8.8hz),6.69-6.70(1h,m),4.79-4.82(4h,m)
6)化合物rm-3的合成(4-(6,7-二氟-2-(甲基丙烯酰氧基甲基)苯并呋喃-5-基)甲基丙烯酸苯酯)
将化合物22(4-(6,7-二氟-2-(羟甲基)苯并呋喃-5-基)苯酚,3.2g,11.6mmol)溶解于无水thf(96.0ml,30ml/g)后,在0℃下将dmap(0.142g,1.2mmol)、tea(5.6ml,40.5mmol)、甲基丙烯酰氯(methacryloylchloride,3.4ml,34.8mmol)按顺序加入,再升温至常温反应1小时。反应结束后,反应物中加入饱和碳酸氢钠溶液,然后分离出有机层。对有机层用无水硫酸钠进行干燥及过滤并减压浓缩。将浓缩残渣用硅胶柱层析法(己烷:etoac=10:1)进行纯化得到白色固体化合物rm-3(1.2g,收率25%)。rm-3的nmr光谱示于图3中。
1h-nmr(cdcl3,varian400mhz):δ7.55(2h,d,j=8.0hz),7.33(1h,d,j=6.4hz),7.23(2h,d,j=8.0hz),6.83(1h,d,j=2.4hz),6.39(1h,s),6.19(1h,s),5.79(1h,s),5.64(1h,s),5.30(2h,s),2.09(3h,s),1.98(3h,s)
合成例4:反应性液晶原的制备(制备rm-4)
1)化合物24的合成(1-(苄氧基)-4-(2,2-二甲氧基乙氧基)苯)
将4-苄氧基苯酚23(4-(benzyloxy)phenol,30.0g,149.8mmol)溶解于dmso(90.0ml,3ml/g)后,再加入koh(10.1g,179.8mmol)。反应溶液中加入溴代乙醛缩二甲醇2(bromoacetaldehyde-dimethylacetal,27.88g,164.8mmol)后,回流反应1小时。反应结束后,冷却至常温,再加入水结束反应。利用过量乙酸乙酯(etoac)进行提取,将有机层用盐水(brine)清洗,然后用无水硫酸钠(anhydrousna2so4)进行干燥及过滤并减压浓缩。将浓缩液用硅胶柱层析法(己烷:etoac=5:1)进行纯化得到无色液体化合物24(40.0g,收率93%)。
1h-nmr(cdcl3,varian400mhz):δ7.32-7.43(5h,m),6.88(4h,q,j=16.8hz),5.01(2h,s),4.70(1h,t,j=5.2hz),3.96(2h,d,j=5.2hz),3.45(6h,s)
2)化合物25的合成(5-(苄氧基)苯并呋喃)
反应容器中加入多聚磷酸(polyphosphoricacid,49.0g,1g/g)、氯苯(980.0ml,20ml/g)用机械搅拌器搅拌均匀。将化合物24(1-(苄氧基)-4-(2,2-二甲氧基乙氧基)苯,56.8g,196.9mmol)溶解于氯苯(735.0ml,15ml/g)后加入反应容器,并在95℃(溶液温度)下反应2小时。将溶液冷却至常温后,利用2n的naoh水溶液进行中和。用二氯甲烷(dcm)进行提取,将有机层用盐水清洗,然后用无水硫酸钠(anhydrousna2so4)进行干燥及过滤并减压浓缩。浓缩后,将反应物用硅胶柱层析法(己烷:ea=20:1)进行纯化得到米色固体化合物25(5.3g,收率12%)。
1h-nmr(cdcl3,varian400mhz):δ7.59(1h,d,j=1.6hz),7.47(2h,d,j=7.2hz),7.38-7.42(3h,m),7.32-7.35(1h,m),7.14(1h,d,j=2.4hz),6.99(1h,dd,j=8.4,2.4hz),6.70(1h,d,j=1.2hz),5.10(2h,s)
3)化合物26的合成((5-(苄氧基)苯并呋喃-2-基)甲醇)
将化合物25(5-(苄氧基)苯并呋喃,7.8g,34.8mmol)、thf(78.0ml,10ml/g)加入溶解后,在氮气环境下冷却至-78℃。在-78℃下缓慢滴入二异丙基胺基锂(43.7ml,87.0mmol,2.0m的thf/庚烷/乙苯溶液),并搅拌30分钟。混合溶液中加入多聚甲醛(p-formaldehyde,7.7g,256.8mmol)后,在常温下反应3小时。反应后,加入饱和氯化铵溶液结束反应。用乙酸乙酯进行提取,将有机层用盐水清洗,然后用无水硫酸钠进行干燥及过滤并减压浓缩。将浓缩液用硅胶柱层析法(己烷:etoac=10:1)进行纯化得到白色固体化合物26(4.9g,收率56%)。
1h-nmr(cdcl3,varian400mhz):δ7.45(2h,d,j=2.4hz),7.31-7.41(4h,m),7.08(1h,d,j=2.4hz),6.96(1h,dd,j=9.0,2.4hz),6.60(1h,s),5.09(2h,s),4.75(2h,d,j=6.0hz),1.86(1h,t,j=6.4hz)
4)化合物27的合成(2-(羟甲基)苯并呋喃-5-醇)
将化合物26((5-(苄氧基)苯并呋喃-2-基)甲醇,4.8g,18.9mmol)、甲醇(96ml,20ml/g)加入溶解后,再加入10%的pd/c(0.48g,10wt%)。反应容器中充入氢气后,在常温下反应5小时。反应结束后,利用乙酸乙酯进行硅藻土过滤及减压浓缩。将浓缩液用硅胶柱层析法(己烷:etoac=5:1)进行纯化得到白色固体化合物27(0.8g,收率26%)。
1h-nmr(dmso,varian400mhz):δ9.07(1h,s),7.25(1h,d,j=9.2hz),6.83(1h,d,j=2.0hz),6.64(1h,dd,j=9.0,2.6hz),6.54(1h,s),5.37(1h,t,j=5.8hz),4.46(2h,d,j=6.0hz)
5)化合物rm-4的合成((5-(甲基丙烯酰氧基)-1h-茚-2-基)甲基丙烯酸甲酯)
在反应容器中将化合物27(2-(羟甲基)苯并呋喃-5-醇,1.6g,9.7mmol)溶解于thf(32.0ml,20ml/g)中。在0℃下将dmap(0.12g,1.0mmol)、tea(4.7ml,34.1mmol)、甲基丙烯酰氯(methacryloylchloride,2.9ml,29.2mmol)按顺序加入后,再升温至常温反应2小时。反应后,加入饱和碳酸氢钠溶液结束反应。用乙酸乙酯进行提取,将有机层用盐水清洗,然后用无水硫酸钠进行干燥及过滤并浓缩。
将浓缩液用硅胶柱层析法(己烷:etoac=10:1)进行纯化得到白色固体化合物rm-4(1.5g,收率52%)。rm-4的nmr光谱示于图4中。
合成例5:反应性液晶原的制备(制备rm-5)
1)化合物29的合成(7-氟-5-(4-(四氢-2h-吡喃-2-基氧基)苯基)苯并呋喃)
反应容器中加入化合物28(5-溴-7-氟苯并呋喃,5.0g,23.2mmol)、化合物13(4,4,5,5-四甲基-2-(4-(四氢-2h-吡喃-2-基氧基)苯基)-1,3,2-二氧硼戊环,6.4g,21.0mmol)、甲苯(64.0ml,10ml/g)、甲醇(6.4ml,1ml/g)。将cs2co3(13.7g,42.0mmol)加入后,在氮气环境下加入pd(pph3)4(1.2g,1.1mmol),并在120℃下回流搅拌3小时进行反应。
反应之后,将反应物冷却至常温,并利用乙酸乙酯进行硅藻土过滤,将过滤后的溶液用盐水清洗,然后用无水硫酸钠进行干燥及过滤并减压浓缩。将浓缩液用硅胶柱层析法(己烷:etoac=7:1)进行纯化得到白色固体化合物29(5.2g,收率71.6%)。
1h-nmr(cdcl3,varian400mhz):δ7.61(3h,m),7.60(1h,s),7.24(1h,s),7.03(2h,dd),6.76(1h,d),5.80(1h,t),3.64-3.74(2h,m),1.78-2.04(2h,m),1.64-1.74(2h,m),1.56-1.59(2h,m)
2)化合物30的合成((7-氟-5-(4-(四氢-2h-吡喃-2-基氧基)苯基)苯并呋喃-2-基)甲醇)
在反应容器中将化合物29(7-氟-5-(4-(四氢-2h-吡喃-2-基氧基)苯基)苯并呋喃,5.2g,17.0mmol)溶解于thf(104.0ml,20ml/g)中,并在氮气环境下将溶液冷却至-78℃。
在-78℃下缓慢滴入正丁基锂(10.2ml,25.5mmol,2.5m的己烷溶液),并搅拌30分钟,然后混合溶液中加入多聚甲醛(p-formaldehyde,3.78g,126.0mmol),再升温至常温并搅拌5小时。
反应物中加入饱和氯化铵溶液结束反应,通过加入乙酸乙酯进行层分离,将有机层用盐水清洗,然后用无水硫酸钠进行干燥及过滤并减压浓缩。将浓缩液用硅胶柱层析法(己烷:etoac=2:1)进行纯化得到白色固体化合物30(3.9g,收率68%)。
1h-nmr(cdcl3,varian400mhz):1h-nmr(cdcl3,varian400mhz):δ7.61(2h,dd),7.60(1h,s),7.24(1h,s),7.03(3h,m),5.80(1h,t),5.12(1h,t),4.39(2h,s)3.64-3.74(2h,m),1.78-2.04(2h,m),1.64-1.74(2h,m),1.56-1.59(2h,m)
3)化合物31的合成(4-(7-氟-2-(羟甲基)苯并呋喃-5-基)苯酚)
将化合物30((7-氟-5-(4-(四氢-2h-吡喃-2-基氧基)苯基)苯并呋喃-2-基)甲醇,1.6g,4.7mmol)、甲醇(32.0ml,20ml/g)加入溶解后,再加入对甲苯磺酸(p-toluenesulfonicacidmonohydrate,0.08g,0.4mmol),并在常温下搅拌2小时。加入水结束反应后,再加入乙酸乙酯(50ml)。
通过减压清除甲醇,并利用乙酸乙酯分离出有机层后,将有机层用盐水清洗,再用无水硫酸钠进行干燥及过滤并减压浓缩。通过用最少量的乙酸乙酯溶解后过量加入己烷得到固体的再结晶方法对浓缩液进行纯化得到白色固体化合物31(1.0g,收率83%)。
1h-nmr(cdcl3,varian400mhz):δ9.67(1h,s),7.60(1h,s),7.42(2h,dd),7.24(1h,s),7.03(1h,s),6.83(1h,dd),5.12(1h,t),4.39(2h,s)
4)化合物rm-5的合成(4-(7-氟-2-(甲基丙烯酰氧基甲基)苯并呋喃-5-基)甲基丙烯酸苯酯)
反应容器中加入化合物31(4-(7-氟-2-(羟甲基)苯并呋喃-5-基)苯酚,1.8g,6.9mmol)、无水thf(55.0ml,30ml/g)进行溶解。在0℃下将dmap(0.08g,0.65mmol)、tea(3.2ml,23.0mmol)、甲基丙烯酰氯(1.86ml,19.0mmol)按顺序加入,并升温至常温反应2小时~3小时。
反应物中加入饱和碳酸氢钠溶液结束反应,并用乙酸乙酯进行提取,将有机层用盐水清洗,再用无水硫酸钠进行干燥及过滤并浓缩。将浓缩后的反应物溶解于二氯甲烷(dcm)中并用柱层析法(己烷:etoac=5:1)进行纯化得到白色固体化合物rm-5(1.3g,收率47%)。rm-5的nmr光谱示于图5中。
合成例6:反应性液晶原的制备(制备rm-6)
1)化合物33的合成(2-(4-(苯并[b]噻吩-5-基)苯氧基)四氢-2h-吡喃)
在反应容器中将化合物13(4,4,5,5-四甲基-2-(4-((四氢-2h-吡喃-2-基)氧基)苯基)-1,3,2-二氧硼戊环,20.0g,65.75mmol)和化合物32(5-溴苯并[b]噻吩,15.41g,72.32mmol)加入甲苯(200ml)和甲醇(20ml)的混合溶液中,再加入csco3(42.85g,231.5mmol)。反应溶液中加入pd(pph3)4(3.80g,3.288mmol),回流搅拌3小时。加入饱和氯化铵溶液结束反应。将反应物冷却至常温,并利用乙酸乙酯进行硅藻土过滤,将过滤后的溶液用盐水清洗后,再用无水硫酸钠进行干燥及过滤并减压浓缩。将浓缩液用硅胶柱层析法(己烷:etoac=10:1)进行纯化得到白色固体化合物33(14.05g,收率68.84%)。
1h-nmr(cdcl3,varian400mhz):1h-nmr(cdcl3,varian400mhz):δ8.12(2h,m),7.99(1h,d),7.65(1h,d),7.61(2h,dd),7.42(1h,d),7.03(2h,dd),5.80(1h,t),3.64-3.74(2h,m),1.78-2.04(2h,m),1.64-1.74(2h,m),1.55-1.59(2h,m)
2)化合物34的合成((5-(4-((四氢-2h-吡喃-2-基)氧基)苯基)苯并[b]噻吩-2-基)甲醇)
在反应容器中将化合物33(2-(4-(苯并[b]噻吩-5-基)苯氧基)四氢-2h-吡喃,14.05g,45.26mmol)溶解于thf(140ml)后,在氮气环境下冷却至-78℃进行搅拌。缓慢滴入正丁基锂(36.8ml,58.84mmol,2.5m的己烷溶液),并在-78℃下进行搅拌。混合溶液中加入多聚甲醛(p-formaldehyde,10.1g,334.9mmol)后,升温至常温并搅拌5小时。反应物中加入饱和氯化铵溶液结束反应,加入乙酸乙酯进行提取,将有机层用盐水清洗。有机层用无水硫酸钠进行干燥及过滤并减压浓缩。将浓缩液用硅胶柱层析法(己烷:etoac=3:1)进行纯化得到化合物34(9.29g,收率60.29%)。
1h-nmr(cdcl3,varian400mhz):1h-nmr(cdcl3,varian400mhz):δ8.12(2h,m),7.99(1h,d),7.61(2h,dd),7.59(1h,s),7.03(2h,dd),5.80(1h,t),5.70(1h,t),4.79(2h,d),3.64-3.74(2h,m),1.78-2.04(2h,m),1.64-1.74(2h,m),1.55-1.59(2h,m)
3)化合物35的合成(4-(2-(羟甲基)苯并[b]噻吩-5-基)苯酚)
反应容器中加入化合物34((5-(4-((四氢-2h-吡喃-2-基)氧基)苯基)苯并[b]噻吩-2-基)甲醇,4.15g,12.19mmol)、甲醇(40.0ml)进行溶解。加入对甲苯磺酸(p-toluenesulfonicacidmonohydrate,0.12g,0.6mmol)后,在常温下搅拌3小时。加水结束反应,然后加入乙酸乙酯进行提取。有机层用无水硫酸钠进行干燥及过滤并减压浓缩。将浓缩液用硅胶柱层析法(己烷:thf=10:1)进行纯化得到化合物35(2.98g,收率95.37%)。
1h-nmr(cdcl3,varian400mhz):1h-nmr(cdcl3,varian400mhz):δ9.67(1h,s),8.12(2h,m),7.99(1h,d),7.59(1h,s),7.42(2h,dd),6.83(2h,dd),5.70(1h,t),4.79(2h,s)
4)化合物rm-6的合成(4-(2-((甲基丙烯酰氧基)甲基)苯并[b]噻吩-5-基)甲基丙烯酸苯酯)
将化合物35(4-(2-(羟甲基)苯并[b]噻吩-5-基)苯酚,2.98g,11.63mmol)溶解于无水thf中。在0℃下将dmap(0.14g,11.63mmol)、tea(5.6ml,40.71mmol)、甲基丙烯酰氯(methacryloylchloride,3.4ml,34.88mmol)按顺序加入,然后升温至常温反应过夜。反应后,加入饱和碳酸氢钠溶液结束反应,并利用乙酸乙酯进行层分离,将有机层用盐水清洗后,再将清洗的有机层用无水硫酸钠进行干燥及过滤并浓缩。将浓缩后的反应物用硅胶柱层析法(己烷:thf=10:1)进行纯化。利用乙酸乙酯和己烷通过再结晶方法进一步纯化得到rm-6(2.19g,47.98%)。rm-6的nmr光谱示于图6中。
试验例1:评价反应性液晶原的光吸收特性
将所述合成例1至6中制备的rm-1至rm-6的光吸收特性与公知的反应性液晶原进行比较。作为公知的反应性液晶原使用了欧洲专利公开ep1498468a1中公开的由下述化学式p-1表示的化合物。
[化学式p-1]
丙烯腈(acn)溶剂中分别加入合成例1至6中制备的rm-1至rm-6和所述由化学式p-1表示的反应性液晶原,准备浓度为0.025mm的四种溶液。然后,对所述每个溶液测定基于波长的吸光度,从而获得uv吸收光谱。所述uv吸收光谱示于图7中。
参照图7,在242nm观察到合成例1中制备的rm-1的最大吸收波长,在240nm观察到合成例2中制备的rm-2的最大吸收波长,在235nm观察到合成例3中制备地rm-3的最大吸收波长。另外,在247nm观察到合成例4中制备的rm-4的最大吸收波长,在235nm观察到合成例5中制备的rm-5的最大吸收波长,在252nm观察到合成例6中制备的rm-6的最大吸收波长。相比之下,在256nm观察到所述由化学式p-1表示的化合物(图7中用比较例1表示)的最大吸收波长。
此外,合成例1至6中制备的rm-1至rm-6显示出高于公知的反应性液晶原的吸光度。因此,可以期待利用根据本发明的反应性液晶原就能提供曝光效率优异的液晶组合物。
实施例1至9及比较例1:液晶组合物的制备
实施例及比较例中使用的液晶化合物用代码来表示。所述代码通过以下方法编写:将构成液晶化合物中心基团的环的符号从左依次填写,并将连接所述中心基团的环的连接基团按顺序填写后,将末端基团填写在右侧。此时,中心基团的环和连接中心基团的环的连接基团之间没有其他区分标记,但中心基团和末端基团之间写入“-”用以区分,而两末端基团用“.”来区分。物质的个别简化符号(代码)整理于下表1中。
【表1】
参见上表1,以下代码表示如下所示结构的液晶化合物。
baf-3.o2:
bb-3.v:
bb-3.u1:
实施例1至9、比较例1及2中使用具有下表2所示组分及物性的主体液晶。
【表2】
低温稳定性:将准备评价低温稳定性的液晶组合物分别取10ml注入小瓶中,然后在低温(-30℃)下保管30天。另外,从保管第一天起每隔24小时观察液晶组合物的状态,以确认是否生成结晶或发生相变。如果30天之前生成结晶或发生相变,就表示为“ng”并记录经过的天数,如果30天后也同样保持初始室温下显示出的液晶相,就表示为“30天”。
tn-i(相变温度):将液晶组合物用滴管在载玻片上滴下一滴后,再盖上盖玻片,以制作样品。向具有温度控制器(mettlertoledofp90)的仪器中放入所述样品,用fp82ht加热台(hotstage)以3℃/min的速度提升温度,并观察样品的变化。记录样品由向列型液晶相变成各向同性液体的时间点的温度,这样的操作反复进行3次以得出平均值。然后,将该值规定为液晶组合物的由向列型液晶相变成各向同性液体的相变温度(tn-i)。
δn(折射率各向异性):液晶组合物的折射率各向异性是在20℃下使用波长为589nm的光线通过目镜上安装偏光片的阿贝折射计进行测定。将主棱镜的表面朝一个方向摩擦(rubbing)后,将液晶组合物滴在主棱镜上。然后,测定了偏光方向与摩擦方向平行时的折射率以及偏光方向与摩擦方向垂直时的折射率。在所述不同的两个方向的折射率中,将更大的折射率规定为ne(extraordinary,非寻常),将更小的折射率规定为no(ordinary,寻常),并将从所述ne减去no的值规定为△n。
δε(介电常数各向异性):液晶组合物的介电常数各向异性是将如下测定的ε∥及ε⊥代入式1中进行计算所得。
[式1]
δε=ε∥-ε⊥
①介电常数ε∥的测定:在两片玻璃基板的形成有ito图案的面上涂布垂直取向剂以形成垂直取向膜。接着,在两片玻璃基板中的任意一个基板上涂布间隔物(spacer)后接合两片玻璃基板,以使垂直取向膜彼此相对且两片玻璃基板之间的间隔(单元间隙)成为4μm。然后,向该元件注入液晶组合物,并用紫外线固化的粘接剂进行密封。之后,用于安捷伦(agilent)制造的4294a型号设备中测定了该元件在20℃下的介电常数ε∥。
②介电常数ε⊥的测定:在两片玻璃基板的形成有ito图案的面上涂布水平取向剂以形成水平取向膜。接着,在两片玻璃基板中的任意一个基板上涂布间隔物(spacer)后接合两片玻璃基板,以使水平取向膜彼此相对且两片玻璃基板之间的间隔(单元间隙)成为4μm。然后,向该元件注入液晶组合物,并用紫外线固化的粘接剂进行密封。之后,用于安捷伦(agilent)制造的4294a型号设备中测定了该元件在20℃下的介电常数ε⊥。
η(粘度,单位:mpa·s):为了测定粘度利用了肖特公司(schottcorp.)的ct52型号设备,其中安装对2ml的体积可测定粘度的毛细管粘度计(capillaryviscometer)后注入待测液晶组合物2ml。然后,使液晶组合物在20℃下稳定30分钟,再使用橡胶滴管将液晶组合物引到测定部位。接着,通过计时器测定液晶组合物流下的速度,由此求出粘度。
γ(旋转粘度,单位:mpa·s):在两片玻璃基板的形成有ito图案的面上涂布垂直取向剂以形成垂直取向膜。接着,在两片玻璃基板中的任意一个基板上涂布间隔物后接合两片玻璃基板,以使垂直取向膜彼此相对且两片玻璃基板之间的间隔(单元间隙)成为50μm。然后,向该元件注入液晶组合物,并用紫外线固化的粘接剂进行密封。之后,使用安装有爱斯佩克公司(especcorp.)制造的温度控制器(su-241型号)的东洋公司(toyocorp.)的6254型号设备测定了该元件在20℃下的旋转粘度。
k11、k33(弹性系数):准备与为了测定所述旋转粘度而制备的元件相同的元件。东洋公司(toyocorp.)的ec1设备中输入单元间隙和介电常数值,向所述元件施加0v至20v的电压通过电容变化来测定弹性系数。
vth(阈值电压;thresholdvoltage):阈值电压是指在基于施加电压的透射率曲线图中,施加电压渐渐增加时元件的透射率变化的时刻的电压,主要在v10(相对于最大透射率达到10%的时刻的电压)前后出现。与所述弹性系数测定相同,准备与为了测定旋转粘度而制备的元件相同的元件,并利用东洋公司(toyocorp.)的ec1设备测定了vth。
按照下表3所示的比例混合主体液晶和反应性液晶原制备出液晶组合物。
【表3】
(单位:重量%)
所述rm-1至rm-6分别是所述合成例1至6中制备的反应性液晶原,所述化合物p-1是所述由化学式p-1表示的反应性液晶原。
试验例2:评价液晶组合物的特性
(1)预倾角(pretiltangle)
将形成有电极和va-聚酰亚胺取向膜的两个基板排列成所述取向膜的摩擦方向不平行(antiparallel)并接合成具有4μm的单元间隙,然后分别注入实施例1至6及比较例1的液晶组合物制作成va-测试单元。
然后,向每个va-测试单元施加10v的电压(交流电压,1khz),并以3.0mw/cm2的强度照射紫外线,以引发va-测试单元内所包含的反应性液晶原的聚合。
此时,按照表4所示改变曝光时间,根据基于晶体旋转法的偏光透射率实验(sesimphotonicstechnology,pams-200)测定紫外线照射后的预倾角(pretiltangle),并示于表4及表5和图8及图9中。
【表4】
(单位:°)
【表5】
(单位:°)
参照上表4及表5和图8及图9,当使用本发明的一个实施方案的反应性液晶原(rm-1至rm-3)时,与使用现有反应性液晶原(化合物p-1)时相比,预倾角更快发生变化。因此,本发明的一个实施方案的反应性液晶原通过较少的曝光量快速聚合。
(2)响应时间
将图案化的电极上形成有va-聚酰亚胺取向膜的两个基板排列成上板的狭缝(slit)位于下板的电极中间并接合成具有3μm的单元间隙,然后分别注入实施例1至6和比较例1及2的液晶组合物制作成pva-测试单元。
然后,向每个pva-测试单元施加3v的电压(交流电压,1khz),并以3.0mw/cm2的强度照射紫外线,以引发pva-测试单元内所包含的反应性液晶原的聚合。
为了测定响应速度,将pva-测试单元以黑色及白色状态驱动,并利用jinyphotonics股份公司的jinyeoscope-050设备测定了pva-测试单元的亮度特性。将所测定的亮度特性转换成电波,由所述电波测定出透射率从10%(黑色)增加到90%(白色)的变动时间(risingtime,ton)以及透射率从90%(白色)减少到10%(黑色)的变动时间(fallingtime,toff)。响应时间(ttotal)是ton和toff的合计值。所测定的响应时间示于表6和表7中。
【表6】
(单位:ms)
【表7】
(单位:ms)
参照所述表6和表7,实施例1至6与比较例1和2相比,基于曝光时间的响应时间的减少速度非常快。这种结果归因于本发明的一个实施方案的反应性液晶原的高吸光度特性,也对应于所述表4和表5所示的基于曝光时间的预倾角变化结果。
因此,当使用本发明的一个实施方案的反应性液晶原时,由于曝光效率优异,可以减少液晶显示装置或光学膜等的制备时间及费用,可以减少长时间暴露于紫外线所导致的各种应力(stress),而且减少残留在液晶组合物中的未反应的反应性液晶原的含量,从而可以提高液晶显示装置的可靠性。
(3)电压保持率(voltageholdingratio,vhr)
将形成有电极和tn-聚酰亚胺取向膜的两个基板排列成所述取向膜的摩擦方向呈90°并接合成具有4μm的单元间隙,然后分别注入实施例1至3、实施例7至9及比较例1的液晶组合物制作成vhr-测试单元。
然后,向每个vhr-测试单元施加10v的电压(交流电压,1khz),并以3.0mw/cm2的强度照射紫外线,以引发vhr-测试单元内所包含的反应性液晶原的聚合。
此时,按照表8和表9所示改变曝光时间测定了电压保持率。所述电压保持率利用液晶电特性设备(toyocorp.,6254型号)在20℃或60℃下向所述vhr-测试单元施加1v的电压60μs并以60hz单位进行测定。测定结果示于下表8和下表9中。
【表8】
(单位:%)
【表9】
(单位:%)
大体而言,包含吸光度高的化合物的液晶组合物与主要包含吸光度低的化合物的液晶组合物相比,存在显示相对低的电压保持率的倾向。然而,实施例1至3显示出高吸光度特性,而且显示出与比较例1同等水平的电压保持率。此外,实施例7至9也有与实施例1至3类似的倾向。
因此,本发明的一个实施方案的反应性液晶原不会导致液晶显示装置的诸多物性变差并显示出优异的曝光效率,从而减少工艺时间及费用,可以提高液晶显示装置的可靠性。
- 还没有人留言评论。精彩留言会获得点赞!