一种mRNA3’末端混合建库的反转录引物及其用途的制作方法
本发明涉及一种mrna3’末端混合建库的反转录引物及其用途。
背景技术
随着新一代高通量测序技术的快速发展,转录组测序(rna-seq)已经成为研究转录组与基因表达的重要手段。然而,常规转录组研究技术主要存在以下问题:基因表达定量不准确、建库和测序价格高。
为了解决上述问题,目前已经开发了mrna3’末端测序技术,该技术提高了基因表达定量的敏感性和准确性。目前lexogen公司的mrna3’末端建库试剂盒最多能混合96个样本(lexogen,https://www.lexogen.com/quantseq-3mrna-sequencing/#quantseqworkflow),可以进行96个样本的自动化建库,该试剂盒省去了mrna分离的复杂操作,前期操作简单成本低,但后期96个样本需要平行独立进行建库操作,而且该产品只有96个双端index,上机时每条lane最多只能混合96个样本,不适合目前novoseq测序仪一条lane需要混合大量样本产出800g以上数据的需求,不适合大规模群体文库构建。
为了进一步提高混合建库的通量和降低文库构建成本,2017年,bush等(plate-seqforgenome-wideregulatorynetworkanalysisofhigh-throughputscreens,bush,e.cetal.,naturecommunications,8,doi:10.1038/s41467-017-00136-z,2017)建立了转录组表达合并文库扩增(plate-seq)技术,该技术使建库工作量大大降低,可在一定程度上降低建库成本,然而,plate-seq文库进行测序时,测序读段(reads)r1端样本标签barcode序列以后的碱基质量很差,基本无法用于后续的数据分析。为避免数据浪费,bush等只将测序读段(reads)r1端测了26bp以读取barcode序列区分样本,后续分析全部使用测序读段(reads)r2端序列。因此,该方法构建的文库只能使用定制化的测序流程进行测序,如果采用目前反应次数固定的测序试剂盒,进行标准的双端150/100bp测序,测序读段(reads)r1端除barcode以外的碱基基本不能用于后续分析。
此外,本领域已知,lexogen的mrna3’末端测序试剂盒市场价格约为260元/样本,plate-seq建库成本约为200元/样本,与常规转录组文库构建价格相比,尽管这两种建库方法的建库成本已经显著降低,但对于大量群体样本的高通量文库构建来讲,这两种建库方法的成本仍然偏高。
因此,对于基因表达定量准确并且建库成本低通量高的建库技术,目前在本领域中还存在着需求。
技术实现要素:
为了解决上述问题,本发明提供了一种mrna3’末端混合建库的反转录引物以及混合建库的方法。
根据本发明的一个目的,提供了一种mrna3’末端混合建库的反转录引物,所述反转录引物包含vn碱基。
在一个实施方式中,所述反转录引物为96个mrna的反转录引物。
在一个实施方式中,所述反转录引物选自表1所示的反转录引物:
表1
根据本发明的另一个目的,提供了一种mrna3’末端混合建库的方法,所述方法包括使用本发明的反转录引物对总rna进行反转录。
在一个实施方式中,混合建库的方法还可包括以下步骤:将反转录完毕的96个样本混合至一个管中,然后降解模板mrna,得到反转录的产物。
在一个实施方式中,混合建库的方法还可包括以下步骤:对反转录的产物进行纯化,并在纯化完成后加入二链合成酶进行二链合成。
在一个实施方式中,混合建库的方法还可包括以下步骤:进行文库片段大小选择,以回收长度150-600bp的片段。
在一个实施方式中,混合建库的方法还可包括以下步骤:进行pcr扩增,以富集模板dna。
在一个实施方式中,混合建库的方法还可包括以下步骤:用等体积的beckmanagencourtampurexpbeads纯化pcr产物,获得mrna3’末端混合文库。
根据本发明的另一个目的,提供了一种mrna3’末端混合建库的方法,所述方法包括以下步骤:
使用权利要求1至3中的任一项所述的反转录引物对总rna进行反转录;
将反转录完毕的96个样本混合至一个管中,然后降解模板mrna,得到反转录的产物;
对反转录的产物进行纯化,并在纯化完成后加入二链合成酶进行二链合成;
进行文库片段大小选择,以回收150-600bp的片段;
进行pcr扩增,以富集模板dna;
用等体积的beckmanagencourtampurexpbeads纯化pcr产物,获得mrna3’末端混合建库。
根据本发明的另一个目的,提供了一种使用本发明的反转录引物进行mrna3’末端混合建库的用途。
在一个实施方式中,所述的混合建库是mrna3’末端混合建库。
使用本发明的反转录引物能够带来以下有益的技术效果:
1)本发明以总rna为起始直接进行反转录,省去了plate-seq建库技术的mrna分离的复杂操作流程,降低了建库成本;
2)反转录时加入锚定碱基,保证从poly(a)的起始位点进行反转录,降低了测序读段(reads)中poly(a)碱基被连续测序的比例,显著提高了测序数据的质量;
3)操作简单,成本显著降低,成本降低至常规转录组的10%以下,建库成本约50元/样本;
4)配套测序平台novoseq、匹配相应的软件开发,建立了自动化、标准化、均一化、简便化的技术体系,使该项技术能够大规模广泛应用在各种动物和植物中,以满足高通量自动化群体转录组文库构建的要求。
附图说明
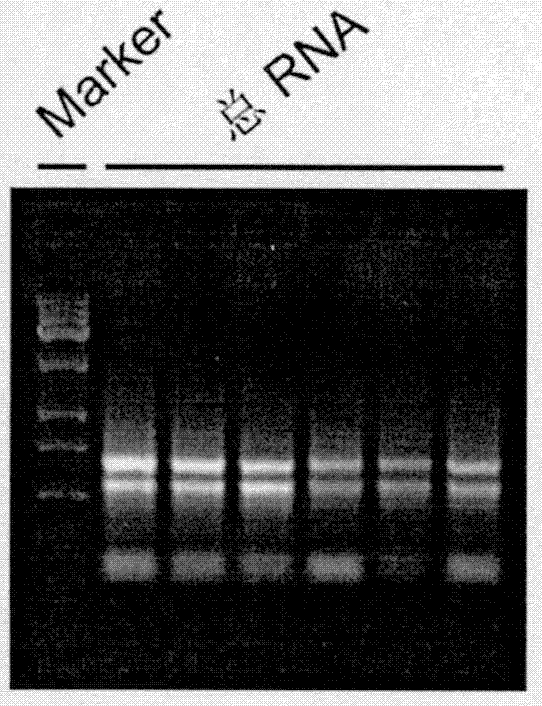
图1为总rna琼脂糖凝胶电泳图,其中marker是指全式金trans15kdnamarker(货号:bm161-01),总rna为提取的植物总rna(例如,从玉米叶片中提取的总rna)。
图2为使用agilent2100检测的本发明建库片段长度分布图。
图3为使用agilent2100检测的对比实施例1建库片段长度分布图,其中对比实施例1与本发明的区别仅在于其反转录引物未加入锚定碱基(即vn碱基),而本发明的反转录引物加入锚定碱基(即vn碱基)。
图4为使用agilent2100检测的对比实施例2(参照plate-seq文献方法,即plate-seqforgenome-wideregulatorynetworkanalysisofhigh-throughputscreens,bush,e.cetal.,naturecommunications,8,doi:10.1038/s41467-017-00136-z,2017)建库片段长度分布图。
图5为本发明和对比实施例1(参见上文描述)构建文库的t碱基频率分布图。
图6为本发明和对比实施例2(参见上文描述)构建文库的t碱基频率分布图。
图7为本发明和对比实施例1(参见上文描述)构建文库的检测基因数目,图7中的重叠基因是对比实施例1和本发明构建文库检测到的相同基因。
图8为本发明和对比实施例2(参见上文描述)构建文库的检测基因数目,图8中的重叠基因是对比实施例2和本发明建库检测到的相同基因。
具体实施方式
下面结合附图和具体实施方式进一步阐述本发明的技术特点。应注意的是,以下具体实施方式仅是示例性的,其不应视为是对本发明的限制。
本发明mrna3’末端混合建库的反转录引物包含vn碱基;优选地,所述反转录引物为96个mrna的反转录引物;更优选地,所述反转录引物选自上述表1所示的反转录引物。
本发明所述的建库方法包括如下步骤:
使用本发明的反转录引物对总rna进行反转录;
将反转录完毕的96个样本混合至一个管中,然后降解模板mrna,得到反转录的产物;
对反转录的产物进行纯化,并在纯化完成后加入二链合成酶进行二链合成;
进行文库片段大小选择,以回收150-600bp的片段;
进行pcr扩增,以富集模板dna;
用等体积的beckmanagencourtampurexpbeads纯化pcr产物,获得mrna3’末端混合文库。
实施例
材料:
取温室培养的土培材料(玉米),生长50天时,取顶部倒2叶,用zymo的direct-zoltmrnaminiprepplus试剂提取总rna,采用琼脂糖凝胶电泳检测提取效果,如图1所示,rna条带清晰,没有明显的降解,可以用于后续的建库操作中。
rna-seq3’末端文库构建流程:
合成二链合成引物以及反转录的锚定引物(所述合成由invitrogen公司进行),然后用depc水稀释到100μm。
本发明文库构建流程:
以总rna为起始,以本发明改进的反转录引物进行反转录和文库构建,流程如下:
(1)反转录:
以总rna为起始,不进行mrna的分离,进行如下操作,使反转录引物和mrna互补结合;
取rnase/dnasefree的200μlpcr管,加入浓度100μm的反转录引物3μl,总rna7μl,混匀离心,放置于pcr仪上,运行94℃2min,迅速置于冰上,离心。
加入以下试剂,进行mrna的反转录:加入0.5mmdntp,10mmdtt,35.8uprotoscriptiireversetranscriptase(neb),轻混离心,放置于pcr仪上,运行25℃5min,42℃1h。cdna可以于-20℃冰箱保存。
(2)降解模板mrna:
加入1μl4×exonucleasei(neb),放置于pcr仪上,运行25℃1h;
加入20μl体积比为1∶1的naoh(1m)和edta(0.5m)的混合物,放置于pcr仪上,运行65℃15min;
加入6m盐酸中和。
(3)使用qiagenminelutepcrpurificationkit进行纯化(货号:28004)具体操作按照产品说明进行,用16μl超纯水洗脱。
(4)合成cdna的互补链:
加1μl10mm的dntp和5μl100μm二链合成引物,放置于pcr仪上,运行70℃2min,迅速放置于冰上5min;
加1μlklenowlargefragmentdnapolymerase(neb),放置于pcr仪上,运行37℃30min;
加入edta直至cdna为50μm时终止反应。
(5)片段选择:
配置2%的琼脂糖凝胶,进行电泳,切取长度150-600bp的片段,使用qiagenminelutegelextractionkit(货号:28604)进行dna回收和纯化。
(6)pcr扩增:
将纯化产物加入nebphusion高保真酶和0.5μmilluminarp1primer和illuminaindexprimer,放置于pcr仪上,进行pcr扩增,其中扩增条件为:98℃30s;98℃,15s;62℃,15s;72℃,60s,运行10-12个循环;72℃,7min;4℃,保持。
(7)pcr产物纯化:
使用等体积的beckmanagencourtampurexpbeads(货号:a63881)进行pcr产物纯化,具体流程参照产品说明进行,最后用22μl超纯水溶,吸上清,获得文库模板dna。
对比实施例1
使用表2的未加锚定碱基(即没有vn碱基)的反转录引物进行反转录,其中文库构建实验流程与本发明上述的文库构建流程相同。
表2
对比实施例2
使用表2的未加锚定碱基(即vn碱基)的反转录引物(参见)进行反转录,其中文库构建流程参照bush文献报道(plate-seqforgenome-wideregulatorynetworkanalysisofhigh-throughputscreens,bush,e.cetal.,naturecommunications,8,doi:10.1038/s41467-017-00136-z,2017)的方法。
对上述文库进行质量检测,文库质量合格后,在illumina测序平台hiseqx10进行pe150双端测序,每个文库测序数据量0.5gb以上。文库下机数据进行过滤,去掉接头序列和低质量碱基,根据样本不同barcode标签分离样本数据,用staralignerversion2.5.3a2(star:ultrafastuniversalrna-seqaligner,dobin,a.,etal.,bioinformatics,29(1),15-21,2012)将reads比对到玉米参考基因组(agpv4)。
结果与分析
plate-seq建库技术首先需要进行mrna的分离,以mrna为起始进行反转录,反转录引物3’端为oligo(t),可以与poly(a)尾不同位点随机结合起始反转录,导致文库插入片段一端含有大量的t碱基,引起测序时碱基质量的下降导致数据不可用。
由图5可以看出,本发明通过加入锚定碱基(即vn碱基),显著降低了测序读段(reads)中t碱基的比例。对比实施例1中未加入锚定碱基时,测序读段(reads)中长度20~30个连续t碱基的频率为63%,相比之下,本发明加入锚定碱基后,测序读段(reads)中长度20~30个连续t碱基的频率降低为8%,69%的测序读段(reads)中连续t碱基长度降低为20以下。
由图6可以看出,与对比实施例2(bush文献报道的plate-seq方法)相比(其中测序读段(reads)中长度20~30个连续t碱基的频率为67%),本发明也显著降低了测序读段(reads)中t碱基的比例。
以上结果说明本发明的加入锚定碱基(即vn碱基)后的反转录引物能显著降低测序读段(reads)中t碱基的比例。
此外,对比分析本发明和对比实施例1、2的检测基因的数目,结果表明(参见图7、图8),使用本发明的反转录引物和方法,在不同测序数据量条件下,检测基因数目平均提高20%以上。
综上所述,以上结果表明,本发明通过对反转录引物进行重新设计并对实验流程进行改进,取得了以下有益效果:①实现了直接以总rna为起始进行文库构建,简化了mrna3’末端混合建库的流程,降低了建库成本,建库成本约50元/样本,远低于plate-seq建库成本(约为200元/样本)和lexogen试剂盒的成本(约为260元/样本);②应用本发明改进的实验流程,显著提高了文库测序数据的质量;③相同测序数据量条件下,与plate-seq方法相比,显著提高了检测基因的数目,提高了基因检测的灵敏度。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种杂环天冬氨基蛋白酶抑制剂的b晶型及其制备方法
- 一种建鲤反转录转座子及转基因载体的构建方法和用图
- 一种杂环天冬氨基蛋白酶抑制剂的c晶型及其制备方法
- 转录谱印记预测药物ms-275在制备小鼠炎症性肠病药物中的应用
- 一种杂环天冬氨基蛋白酶抑制剂的a晶型及其制备方法
- 嗜热链球菌CRISPR-Cas9系统识别的人CCR5基因的靶序列和sgRNA及其应用
- 一种治疗艾滋病的中药及其制备方法
- 脑膜炎双球菌CRISPR-Cas9系统识别的人CCR5基因的靶序列和sgRNA及其应用
- 脑膜炎双球菌CRISPR-Cas9系统识别的人CXCR4基因的靶序列和sgRNA及其应用
- 嗜热链球菌CRISPR-Cas9系统识别的人CCR5基因的靶序列和sgRNA及其应用