用于长读长高质量测序的核酸文库的构建方法、测序方法及试剂与流程
本发明涉及测序
技术领域:
,尤其涉及一种用于长读长高质量测序的核酸文库的构建方法、测序方法及试剂。
背景技术:
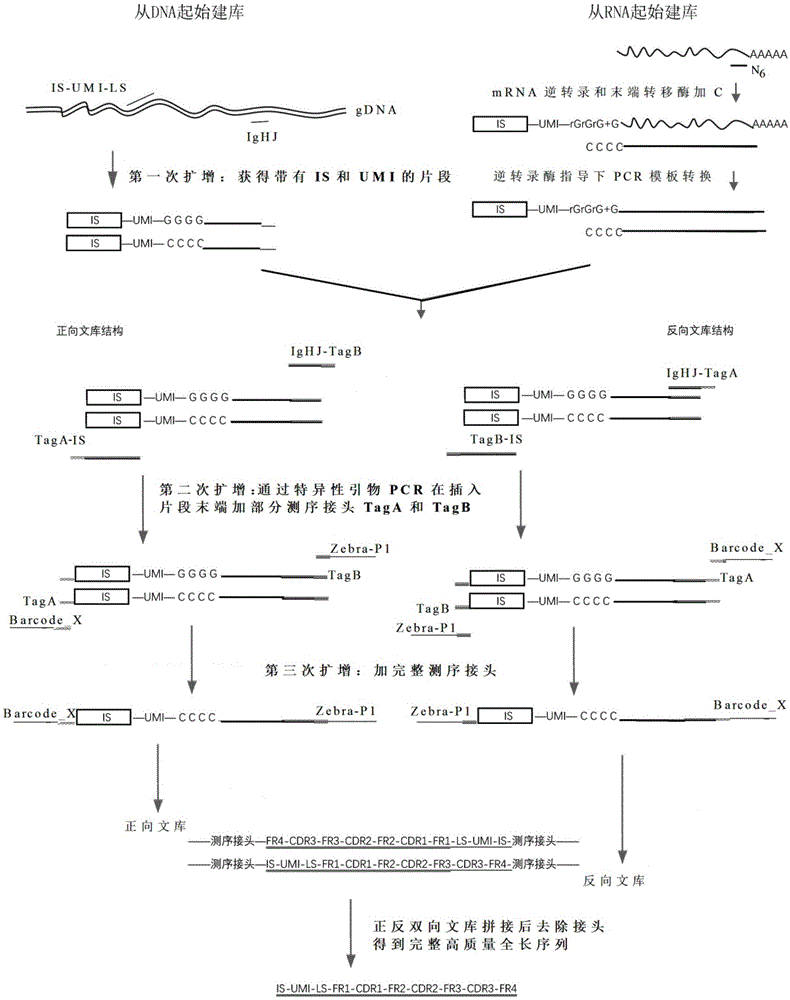
:长pcr产物文库的高通量测序,如16srrna细菌鉴定、基于高通量测序的hla分型以及免疫组库测序等,在科学研究中经常遇到。以免疫组库可变区全长文库(主峰300bp至600bp)为例,一般采用illumina公司的pe250(hiseq)或者pe300(miseq)测序方式。所使用的进口测序仪器和测序试剂价格都很昂贵,购买货期较长,并且保质期较短。同时,对于pcr产物的双末端高通量测序策略而言,pe250或者pe300测序模式下的读长2(reads2)的末端100bp质量值迅速下降(q30从70-80%以上下降至50%以下)。而国产测序仪目前还不能做到对主峰300bp至600bp插入片段序列的高质量测序。对单末端测序来讲,能保证前300bp碱基的高质量值,q30大于等于70%~80%,大于300bp时碱基质量值迅速下降。但是国产测序仪器和试剂价格相对便宜,货期短,因此若能开发出基于国产测序仪的长pcr产物的建库和测序策略,将具有很大竞争优势。技术实现要素:本发明一种用于长读长高质量测序的核酸文库的构建方法、测序方法及试剂。根据本发明的第一方面,本发明提供一种核酸文库的构建方法,该方法包括:(a)以dna或rna作为起始模板材料进行第一次扩增,上述第一次扩增中使用的引物包括正向引物和反向引物,其中上述正向引物由5’端至3’端依次包括一段公共序列、一段唯一分子识别标记序列以及一段与模板结合的序列,上述反向引物是目标特异性序列或非特异性序列;(b)以上述第一次扩增的产物为模板,进行第二次扩增,上述第二次扩增包括在各自独立体系中进行的正向文库扩增和反向文库扩增,上述正向文库扩增中使用的引物包括第一引物和第二引物,上述第一引物由5’端至3’端依次包括部分测序接头序列a和上述公共序列,上述第二引物由5’端至3’端依次包括部分测序接头序列b和目标特异性序列;上述反向文库扩增中使用的引物包括第三引物和第四引物,上述第三引物由5’端至3’端依次包括上述部分测序接头序列b和上述公共序列,上述第四引物由5’端至3’端依次包括上述部分测序接头序列a和上述目标特异性序列;以及(c)以上述正向文库扩增和反向文库扩增的产物为模板,进行第三次扩增,上述第三次扩增中使用的引物包括正向引物和反向引物,上述正向引物包括位于3’端的上述部分测序接头序列a及其上游序列,该上游序列包括用于区分样本的条形码序列,上述反向引物包括位于3’端的上述部分测序接头序列b及其上游序列。优选地,上述起始模板材料是dna,上述第一次扩增使用的正向引物中与模板结合的序列是一段特异性序列,上述第一次扩增使用的反向引物是目标特异性序列。优选地,上述起始模板材料是rna,上述第一次扩增使用的正向引物是模板转换寡核苷酸(tso),上述模板转换寡核苷酸中与模板结合的序列包括位于3’端的锁核酸(lna),上述第一次扩增使用的反向引物是随机引物或oligo-dt引物。优选地,上述模板转换寡核苷酸中与模板结合的序列包括位于3’端的核糖核苷酸残基(rn)和上述锁核酸(lna);更优选地,上述核糖核苷酸残基是核糖鸟嘌呤(rg),上述锁核酸是锁鸟嘌呤(+g),最优选地,上述与模板结合的序列包括位于3’端的rgrgrg+g。优选地,上述核酸文库是pcr产物文库,优选地,pcr产物文库是免疫组库全长文库,更优选地,上述免疫组库全长文库的主峰是300bp至600bp。优选地,上述核酸文库适用于illumina、iontorrent、bgiseq或mgiseq测序平台,更优选bgiseq测序平台。根据本发明的第二方面,本发明提供一种由第一方面的核酸文库的构建方法构建得到的核酸文库。根据本发明的第三方面,本发明提供一种测序方法,该方法包括:根据第一方面的核酸文库的构建方法构建核酸文库;对上述核酸文库进行测序。根据本发明的第四方面,本发明提供一种用于构建核酸文库的引物组合,该引物组合包括:用于以dna或rna作为起始模板材料进行第一次扩增的正向引物,上述正向引物由5’端至3’端依次包括一段公共序列、一段唯一分子识别标记序列以及一段与模板结合的序列,该与模板结合的序列是特异性序列;或上述正向引物是模板转换寡核苷酸(tso),上述模板转换寡核苷酸由5’端至3’端依次包括一段公共序列、一段唯一分子识别标记序列以及一段与模板结合的序列,该与模板结合的序列包括位于3’端的锁核酸(lna)。优选地,上述引物组合还包括:用于以dna或rna作为起始模板材料进行第一次扩增的反向引物,该反向引物是目标特异性序列或非特异性序列。优选地,上述引物组合还包括:用于以上述第一次扩增的产物为模板进行第二次扩增的引物,其包括正向文库扩增引物和反向文库扩增引物,上述正向文库扩增引物包括第一引物和第二引物,上述第一引物由5’端至3’端依次包括部分测序接头序列a和上述公共序列,上述第二引物由5’端至3’端依次包括部分测序接头序列b和目标特异性序列;上述反向文库扩增引物包括第三引物和第四引物,上述第三引物由5’端至3’端依次包括上述部分测序接头序列b和上述公共序列,上述第四引物由5’端至3’端依次包括上述部分测序接头序列a和上述目标特异性序列;以及用于以上述第二次扩增的产物为模板进行第三次扩增的引物,其包括正向引物和反向引物,上述正向引物包括位于3’端的上述部分测序接头序列a及其上游序列,该上游序列包括用于区分样本的条形码序列,上述反向引物包括位于3’端的上述部分测序接头序列b及其上游序列。本发明的建库方法将唯一分子识别标记(umi)技术与pcr产物定向加正反测序接头形成正反双向文库技术结合起来,根据正反双向文库的测序重叠部分拼接出高质量(q30>77%)的主峰(300bp至1000bp,优选300bp至600bp),从而实现长读长测序,其中umi用于同一样本的正反双向文库测序数据拼接。本发明能够显著降低测序成本。此外,本发明对多种平台具有广泛的适用性,且同时适用于单端测序和双端测序策略。附图说明图1为本发明示例性的核酸文库的构建方法流程图以及测序结果拼接原理示意图。图2本发明一个实施例中测序得到的两个正反向文库的结构示意图。具体实施方式下面将更详细地描述本发明的用于长读长高质量测序的核酸文库的构建方法、测序方法及试剂。除非另外定义,否则详述中使用的技术和科学术语具有与本发明领域的技术人员理解相同的含义。本发明中,术语―长读长”是指主峰在300bp以上的文库,例如300bp至1000bp,优选300bp至600bp,这样的核酸文库包括但不限于16srrna细菌鉴定、基于高通量测序的hla分型以及免疫组库测序文库,尤其是免疫组库全长文库,优选地,免疫组库全长文库的主峰是300bp至600bp。本发明中,术语―高质量测序”是指测序质量值q30>77%的测序,优选测序质量值q30>80%的测序。本发明的核酸文库适用于illumina、iontorrent、bgiseq或mgiseq测序平台,更优选bgiseq测序平台。本发明适用于dna或rna或二者的组合作为起始材料的文库构建。图1示出了一个示例性的核酸文库的构建方法流程图以及测序结果拼接原理示意图。在图1中以免疫保守序列ighj的扩增为例。需要说明的是,图1是示例性的,仅是为本发明的原理和方法能够更加直观形象地被理解,因此不能理解为对本发明保护范围的限制。参考图1,一种核酸文库的构建方法,包括如下步骤:(a)以dna或rna作为起始模板材料进行第一次扩增,上述第一次扩增中使用的引物包括正向引物(图1中is-umi-ls或is-umi-rgrgrg+g)和反向引物(图1中ighj或n6随机引物),其中上述正向引物由5’端至3’端依次包括一段公共序列(is)、一段唯一分子识别标记序列(umi)以及一段与模板结合的序列(ls),上述反向引物是目标特异性序列(图1中ighj)或非特异性序列(图1中n6随机引物);(b)以上述第一次扩增的产物为模板,进行第二次扩增,上述第二次扩增包括在各自独立体系中进行的正向文库扩增和反向文库扩增,上述正向文库扩增中使用的引物包括第一引物(图1中taga-is)和第二引物(图1中ighj-tagb),上述第一引物由5’端至3’端依次包括部分测序接头序列a(图1中taga)和上述公共序列(is),上述第二引物由5’端至3’端依次包括部分测序接头序列b(图1中tagb)和目标特异性序列(图1中ighj);上述反向文库扩增中使用的引物包括第三引物(图1中tagb-is)和第四引物(图1中ighj-taga),上述第三引物由5’端至3’端依次包括上述部分测序接头序列b(图1中tagb)和上述公共序列(is),上述第四引物由5’端至3’端依次包括上述部分测序接头序列a(图1中taga)和上述目标特异性序列(图1中ighj);以及(c)以上述正向文库扩增和反向文库扩增的产物为模板,进行第三次扩增,上述第三次扩增中使用的引物包括正向引物(barcode_x)和反向引物(zebra_p1),上述正向引物包括位于3’端的上述部分测序接头序列a(图1中ighj-taga)及其上游序列,该上游序列包括用于区分样本的条形码(barcode)序列,上述反向引物包括位于3’端的上述部分测序接头序列b(图1中tagb)及其上游序列。需要说明的是,在本发明中,所谓―正向”和―反向”仅用于指代核酸两条链的扩增方向,在―正向”表示一条链的扩增方向的情况下,―反向”表示另一条链的扩增方向。相应地,―正向引物”和―反向引物”也应当作类似理解。本发明中,第一次扩增使用的正向引物由5’端至3’端依次包括一段公共序列、一段唯一分子识别标记序列(umi)以及一段与模板结合的序列(ls)。其中,公共序列仅以is为例,可更换为任意一段序列,关键就在于该公共序列也出现在第二次扩增的第一引物和第三引物的3’端,因此能够有效地实现连续扩增。对该公共序列而言,所谓―公共”是指针对所有扩增产物,虽然不同克隆来源具有不同的唯一分子识别标记序列(umi),但是公共序列是一致的。因此,公共序列与umi配合不但能够实现不同克隆来源的片段同步扩增,而且能够区分不同来源的扩增片段。唯一分子识别标记序列(umi)的一种典型但非限定性的例子是nnnnunnnnunnnnu,其中n可以是任意碱基,该umi可更换为任意间断的或不间断的n个核苷酸长度的序列,所谓―间断”是指例如上述例子中n被u间隔开。该umi的作用是标记来源于同一条克隆的pcr产物,即来源于同一条克隆的pcr产物都带有相同的umi。本发明中,在第一次扩增时,反向引物可以是目标特异性序列(如图1中ighj),可以是扩增特定基因的特异性引物,也可以是非特异性序列(如图1中n6随机引物),例如任何随机引物或oligo-dt引物,其中随机引物可以是6碱基的随机引物(6-mer),也可以是其他碱基数量的随机引物,oligo-dt引物即一定数量的t碱基连续序列,可以带有其他碱基或修饰,例如可以是5′-aagcagtggtatcaacgcagagtact30vn-3′序列,其中―n”可以是任何核苷碱基,并且―v”选自下组―a”、―c”以及―g”。理论上,无论起始模板材料是dna还是rna的情况下,第一次扩增时,反向引物都可以是目标特异性序列或非特异性序列。但是,在优选的实施例中,当起始模板材料是dna时,第一次扩增使用的正向引物中与模板结合的序列是一段特异性序列,即该正向引物具有这样的结构:5′-公共序列-唯一分子识别标记序列-特异性序列-3′;同时,第一次扩增使用的反向引物是目标特异性序列。在优选的实施例中,当起始模板材料是rna时,第一次扩增使用的正向引物是模板转换寡核苷酸(template-switchingoligos,tso),该模板转换寡核苷酸中与模板结合的序列包括位于3’端的锁核酸(lna),上述第一次扩增使用的反向引物是随机引物或oligo-dt引物。关于模板转换寡核苷酸的技术,在中国专利申请cn105579587a以及文献(simonepicelli,etal.full-lengthrna-seqfromsinglecellsusingsmart-seq2.natureprotocols.9(1):171–181(2014))中有介绍。简而言之,使cdna合成引物(例如随机引物或oligo-dt引物等)退火于rna分子并且合成第一cdna链以形成rna-cdna中间体;然后,通过使该rna-cdna中间体与模板转换寡核苷酸(tso)在适于第一cdna链延伸的条件下接触进行逆转录酶(例如moloneymurineleukemia(m-mlv)reversetranscriptase)反应。参考图1,在n6随机引物引导下,以rna作为模板进行延伸,当延伸到cdna的3’端时会加上若干个c碱基,例如三个c碱基。然后,模板转换寡核苷酸末端与上述c碱基形成稳定的配对关系,在逆转录酶作用下,第一cdna链继续以模板转换寡核苷酸为模板延伸。需要说明的是,锁核酸(lockednucleicacid,lna)是一种经过修饰的rna,lna中一部分核糖上的2'与4'碳连结在一起。因此,一方面增强了其自身的稳定性,另一方面能够非常强的退火到cdna的3’端的互补碱基上,大大提高逆转率效率。需要说明的是,图1示出的仅是模板转换寡核苷酸(tso)3’端具有核糖鸟嘌呤(rg)和锁鸟嘌呤(+g)的情况,但是在具体应用中,本发明的模板转换寡核苷酸3’端可以包括任何核糖核苷酸残基(rn)和锁核酸(lna),例如锁核酸(lna)残基可以是:锁鸟嘌呤、锁腺嘌呤、锁尿嘧啶、锁胸腺嘧啶、锁胞嘧啶以及锁5-甲基胞嘧啶等。在优选实施例中,模板转换寡核苷酸(tso)3’端具有rgrgrg+g表征的结构。本发明中,第二次扩增使用的第一引物由5’端至3’端依次包括部分测序接头序列a(图1中taga)和公共序列(is),该公共序列即是与第一次扩增使用的正向引物5’端的公共序列相同的序列,而部分测序接头序列a(taga)是指测序平台上的测序接头序列的一部分,该序列因测序平台不同而改变。类似地,第二次扩增使用的第三引物由5’端至3’端依次包括部分测序接头序列b(图1中tagb)和公共序列(is),该公共序列也是与第一次扩增使用的正向引物5’端的公共序列相同的序列,而部分测序接头序列b是指测序平台上的另一个测序接头序列的一部分,该序列也因测序平台不同而改变。在本发明的一个实施例中,测序平台是bgi-seq平台,相应地,taga和tagb分别是gaccgcttggcctccgactt和acatggctacgatccgactt序列,它们分别用于下一步扩增中与完整测序接头引物进行搭桥结合。本发明中,第三次扩增使用的正向引物(图1中barcode_x)和反向引物(图1中zebra_p1),分别是测序平台上的两个完整测序接头引物,分别结合扩增片段的两端,在两端分别加上两个完整的测序接头。其中,正向引物包括位于3’端的部分测序接头序列a(与第二次扩增中的部分测序接头序列a相同)及其上游序列,该上游序列包括用于区分样本的条形码(barcode)序列,该上游序列除了条形码序列以外,还可以包括其他序列,这些其它序列可以介于条形码序列与部分测序接头序列a之间,也可以位于条形码序列的上游5’端,或者兼而有之。反向引物包括位于3’端的部分测序接头序列b(与第二次扩增中的部分测序接头序列b相同)及其上游序列。需要说明,第三次扩增使用的正向引物和反向引物也会因测序平台而定,在本发明的一个实施例中,测序平台是bgi-seq平台,相应地,正向引物是tgtgagccaaggagttgxxxxxxxxxxttgtcttcctaagaccgcttggcctccgactt序列(图1中barcode_x),其中xxxxxxxxxx是条形码序列。反向引物是gaacgacatggctacgatccgactt序列(图1中zebra_p1)。以下通过实施例详细说明本发明的技术方案,应当理解,实施例仅是示例性的,不能理解为对本发明保护范围的限制。i.第一步扩增:目的基因富集:rna样本逆转录加umi或dna样本pcr加umi1.以rna样本作为起始原料,混合rna样本,如下表1所示:表1组分体积rna大于2μgn6(6碱基随机引物,1μg/μl)0.5-1μldepc-水补充到15μl总体积15μl以上样品在65℃孵育7min,冰浴5min。2.加入以下表2中的成分反转录生成第一条cdna链,每条cdna链上标记umi:表2以上体系在25℃孵育10min;然后42℃孵育2h;最后72℃15min。3.以dna样本作为起始原料,配置如下表3所示的反应体系:表3组分体积2×mastermix(neb公司)25μl5×q溶液(neb公司)5μlis-umi-ls引物(10μm)1μlighj引物(10μm)1μldna4μl无核糖核酸酶的水补齐至50μlii.第二步扩增:4.pcr富集目的基因,以ighj重链50μl扩增体系为例,配置如下表4、表5所示的反应体系:表4正向文库组分体积2×mastermix25μl5×q溶液5μltaga-is引物(10μm)1μlighj-tagb引物(10μm)1μl第一步扩增的产物4μl无核糖核酸酶的水补齐至50μl表5pcr反应程序是:95℃15min;94℃30s,65℃90s,72℃30s,10个循环;72℃5min;12℃保温。5.使用ampurexp磁珠纯化两遍将表4和表5的pcr反应产物转移至1.5ml离心管中,用ampurexpdna纯化试剂盒(spri磁珠)纯化扩增后的样品:1)取出4℃保存的ampurexp磁珠,室温放置30min平衡;2)使用前振荡均匀,按照样品体积加入0.5-1.5倍体积磁珠(50μl)并混匀,静置3min,瞬时离心3秒;3)将1.5ml离心管转移放置在磁力架上,静置3min至澄清;4)小心吸去上清,不要触及磁珠(1.5ml离心管放在磁力架上);5)加入500μl75%乙醇,轻轻吹打磁珠2-3次,等待30秒,弃上清(加入乙醇时应缓缓加入,尽量不要让液体往磁珠方向添加,否则会使磁珠脱离管体而损耗);6)重复步骤5),尽量去除上清(此步不需要吹打磁珠);7)置恒温混匀仪37℃干燥2min左右,磁珠表面没有水分即可(仔细观察磁珠情况,避免磁珠干裂之后再持续加热,持续加热有使磁珠崩离加样孔的潜在风险,造成损失和样品间污染,个别未干孔可取离干燥仪静置风干);8)往1.5ml离心管中加入50μl无核酸酶的水,充分混匀,静置5min,然后置于磁力架约5min至澄清;9)将50μl澄清液转移至事先准备好的新的pcr管中;10)重复步骤2)至9);11)往1.5ml离心管中加入24μl无核酸酶的水,充分混匀,静置5min,然后置于磁力架约5min至澄清;12)将23μl澄清液转移至事先准备好的新的pcr管中(转管时需要特别注意将转移至对应管中,避免出错)。iii.第三步扩增:6.测序文库构建:目的基因两端引入完整测序接头以上纯化样品按照下列表6加入pcr反应体系进行扩增,最终得到带有正反接头的重链免疫组库。表6组分体积纯化的dna23μlzebra-p1引物(10μm)1μlbarcode_x引物(10μm)1μlphusiondna酶25μl总体积50μlpcr反应程序是:98℃1min;98℃20s,65℃30s,72℃30s,30个循环;72℃5min;12℃保温。7.使用2%琼脂糖凝胶回收1)配置2%的回收胶;2)将多重pcr产物进行电泳,100v,400ma,电泳2-3h;3)eb染胶(或配置凝胶的时候加入eb替代荧光染料);4)片段选择:重链切胶回收片段范围是400-600bp;5)切胶回收:使用30μl左右无核酸酶的水回溶。以上步骤使用的引物序列如表7所示:表7在以上表7中,正向taga序列(gaccgcttggcctccgactt)和反向tagb序列(acatggctacgatccgactt)属于bgi-seq测序平台上机测序接头的部分序列,用于下一步与完整测序接头引物进行搭桥结合。在以上表7中,barcode_x引物中xxxxxxxxxx为建库时用于区分样本的条形码(barcode)序列,用于测序后拆分数据。is-umi-ls引物为起始样本是dna时的引物,非通用引物,引物结构为―一段公共序列+umi分子标记+特异性引物”,公共序列仅以is为例,可更换为任意一段序列,taga和tagb亦如此,结构中的umi分子标记仅以nnnnunnnnunnnnu为例,可更换为任意间断的或不间断的n个核苷酸长度的序列;而结构中特异性引物,需要根据pcr产物进行设计,也非固定,表7中仅以ls前导区一段序列为例。tso-umi中u为尿嘧啶核糖核苷酸,n为a、t、c、g任意一种碱基类型,rg代表鸟嘌呤核糖核苷酸,+g代表锁鸟嘌呤。将回收得到的产物,使用表7中on4563夹板(splint)序列辅助,在t4dna连接酶作用下进行环化。然后使用核酸外切酶i(exoi)和核酸外切酶iii(exoiii)消化未环化的核酸片段。最后纯化得到环化文库。使用环化文库进行纳米球制备、测序上机和数据分析。纳米球制备和测序上机请参考http://www.seq500.com/。之后下机序列经过imonitor软件的生物信息学分析,基本的分析思路是,将下机的免疫组库序列与国际通用的免疫组库数据库imgt(http://www.imgt.org/vquest/refseqh.html)中人的胚系基因进行分析比对,统计得出该免疫组库的序列信息等。得到下机数据后,进行基本的质量值过滤,根据样本条形码序列将各个样本数据拆开,每个样本都将有正反双向两个文库,再根据umi和正反双向文库的重叠序列区域,将正反双向文库数据拼接成一条完整的高质量值的免疫组库序列(主峰约400bp),基本原则是来源于同一条克隆的pcr产物,它们的umi分子标记是一样的,约有80-150bp的重叠部分反向互补以后也是一样的。来源于同一扩增模板的正反双向文库数据可以利用质量值较高的300bp部分序列将全长500bp拼接出来。同时可以校正80-150bp重叠部分的测序和pcr错误。实验结果:1.生物信息分析rna起始样本文库根据上述技术方案,用健康人rna样本a做测试,建立正反双向免疫组库rbz-tag1和rbz-tag2,分别标记不同的条形码序列。单端se500测序下机数据根据条形码序列拆分文库。图2示出了两个正反向文库的结构,正向文库测得方向是从fr4区到ls区(leadersequence,前导区)的反义链,末端包含umi信息;反向文库测得方向是从ls区到fr4区的正义链,起始端含有umi信息。两个文库的cdr1-fr2区的q30测序质量值都在80%以上,同时也是两个文库的重叠部分,因此可用于拼接。此时将来自于两个文库umi相同且cdr1-fr2区序列相同的两类克隆判定为同一克隆,用于全长拼接,具体来讲,即利用反向文库rbz-tag2的ls-fr1-cdr1-fr2高质量的序列信息和正向文库的cdr1-fr2-cdr2-fr3-cdr3-fr4高质量的序列信息,拼接出完整的高质量的ls-fr1-cdr1-fr2-cdr2-fr3-cdr3-fr4全长序列信息。以测序得到的其中一种umi(acaatgggattgcat)为例来讲:正向文库序列如下:反向文库序列如下:gcgctgctctgatacactcttggagctgagcagtctgagatctgagggcacggccgtggggagacatcaggggcgatttacacagggtgctagctttgactccttgggccagggaatactg通过生物信息分析发现:两个文库前300bp碱基质量值较高(双横线,q30≥80%),后200bp碱基质量值较低(单横线,q30%<70%),其中标记为大写斜体的碱基位点与对应文库前端300bp中标记为大写粗体本应反向互补对应,但因测序错误导致不对应,且大写斜体的碱基位点均位于末端200bp低质量的序列里,根据大写粗体的正确碱基将其校正,拼接得到的全长序列如下:序列分析结果如下:(1)aagcagtggtatcaacgcagagtacaatgggattgcatcttggggg为tso-umi引物,其中aagcagtggtatcaacgcagagt为is序列互补结合位置;acaatgggattgcat为umi标记信息;(2)双横线区域为ls前导区序列;(3)大写字母为抗体全长可变区序列信息,碱基质量q30>80%;(4)波浪下划线区域为正反文库的高质量重叠区域,依靠本区域序列和umi信息共同判定正反文库中来源于同一pcr模板扩增的克隆。在umi数量足够多的情况下,单独依靠umi即可判定。2.生物信息分析dna起始样本文库根据上述技术方案,我们用健康人dna样本yh(人永生化b细胞系——炎黄细胞系gdna,参考doi:10.1038/nature07484.pmid:18987735.)做测试,建立正反双向免疫组库yh-tag1和yh-tag2,分别标记不同的条形码序列。单端se500测序下机数据根据条形码序列拆分后,两个文库分别得到323657条和2993567条读长,去除低质量数据分别得到2800090条和2334080条读长。图2示出了两个正反向文库的结构,正向文库测得方向是从fr4区到ls区(leadersequence,前导区)的反义链,末端包含umi信息;反向文库测得方向是从ls区到fr4区的正义链,起始端含有umi信息。生物信息序列拼接方法同上生物信息分析rna起始样本文库,得到类似结果。本发明的方法将umi技术与pcr产物文库定向加正反测序接头的优点结合了起来,能实现测序平台——尤其是bgi-seq平台长读长测序的目的;本发明的方法开发出了umi能用于拼接正反双向文库的功能;本发明的方法同时显著降低了测序成本50%以上。一般而言,miseqpe300测序一个运行(run)的市场价在2至3万元人民币,hiseqpe250测序一个运行(run)的市场价在5至7万元人民币,而使用该方法的bgi-seq500单端测序的一个运行(run)的市场价只有1至2万元人民币。本发明的方法适用的测序平台包含但不限于illumina、iontorrent、bgiseq、mgiseq等,测序策略包括但不限于单端测序(se50-se1000)和双端测序;pcr产物包括但不限于免疫组库、16srrna细菌鉴定、基于高通量测序的hla分型等任意产物。本发明理论可以拼接出300bp-1000bp的解释。目前国产测序仪bgi-seq平台单端测序长度在500bp,但只有前面300bp质量值较高,后面200bp较低,使用本发明的方法,正反双向文库中同一umi且有重叠部分的对应读长拼接后可以得到质量值较高的550-600bp的序列。随着测序技术的发展,单端测序长度会不断增长至1000bp,而对于pcr产物文库高通量测序而言,前端约2/3的测序质量值比较可信,末端约1/3的测序质量值较低,同样可采用本发明中的正反双向文库建库方法,拼接出质量值较高的全长序列。以上内容是结合具体的实施方式对本发明所作的进一步详细说明,不能认定本发明的具体实施只局限于这些说明。对于本发明所属
技术领域:
的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干简单推演或替换,都应当视为属于本发明的保护范围。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1