一种侧链含高电子迁移率芳杂环的醇/水溶性有机小分子阴极界面材料的制作方法
本发明涉及有机太阳能电池材料技术领域,具体涉及一种侧链含高电子迁移率芳杂环的醇/水溶性有机小分子阴极界面材料的制备方法及其在有机太阳能电池中的应用。
技术背景
工业革命以来,能源的使用呈现几何级别的增加,目前对人类社会发展来说最主要的能源是传统的煤、石油和天然气资源,但是由于传统能源具有不可再生性,严重威胁了人类社会的可持续发展。同时,传统能源的使用也给人类生存环境带来了极大危害,影响了人类的健康和生存。太阳能作为一种取之不尽、用之不竭的可再生能源是人类社会维持可持续发展的必然选择。太阳能电池作为直接将太阳能转化成电能的装置,近年来受到人们的极大关注。尤其是有机太阳能电池,由于其质量轻、柔性、可卷对卷的大规模生产等优点,被认为是下一代太阳能电池的典型代表。在科研工作者的不断努力下,目前基于单层有机太阳能电池的效率已经超过了14%。但是由于在传统有机太阳能电池的制备过程中一般使用活泼金属钙作为阴极夹层,因此对器件的封装提出了极大的挑战。随着阴极界面的发展和醇/水溶性的阴极界面层的使用,有机太阳能电池的稳定性和效率均获得了极大的提升。但是目前使用的醇/水溶性阴极界面材料的导电性能较差,因此在制备过程中对界面层厚度有严格的要求,一般厚度控制在5纳米左右,这样的精度不利于以后的大规模工业生产。本发明提供一类对膜厚不敏感的阴极界面层材料的制备方法,即将噻二唑和噁二唑等具有高电子迁移率的芳杂环单元引入到有机小分子界面材料的侧链中,提高材料的导电性,改善其阴极界面修饰能力,为制备膜厚不敏感的高效阴极界面层提供技术保障。
本发明分别将含有2,5-二苯基-1,3,4-噻二唑与2,5-二苯基-1,3,4-噁二唑柔性侧链的单体,通过suzuki偶联的方法制备了一类醇/水溶性的有机小分子阴极界面层材料,通过这些高电子迁移率结构单元的引入,有效提升了材料的导电性能,制备出对膜厚不敏感的阴极界面层材料,解决了有机太阳能电池不适宜大规模生产的问题。
技术实现要素:
基于以上所述,本发明的目的在于提出一类高电子迁移率的醇/水溶性有机小分子阴极界面材料,并且将其作为阴极界面层应用于有机太阳能电池中。重要的是通过引入了噻二唑/噁二唑等电子传输性能优异的官能团,改善了其膜厚不敏感性,有利于将来有机太阳能电池的湿法大规模工业生产。
本发明的技术方案为:
本发明提供了一类高电子迁移率的醇/水溶性的有机小分子阴极界面材料,该类有机小分子阴极界面材料具有以下通式:
其中,r为末端含有溴化三甲铵基的c1-c12的烷基;a选用式ⅱ中所示的含高电子迁移率的芳杂环官能团,其中r1为c1-c12的烷基或者烷氧基;x为o、s、se或te等杂原子。
优选的方案,r1为c2-c8的烷氧基;x为o或者s原子,a选用式ⅲ和式ⅳ所示的基团结构:
2.如权利要求1所述的有机小分子阴极界面材料,其特征在于r优先选择c2-c8的直链烷基或者烷氧基。
3.如权利要求1所述的有机小分子阴极界面材料,其特征在于x优先选择o和s杂原子。
4.如权利要求1所述的有机小分子阴极界面材料,其特征在于a优先选用下列结构:
其中r1为c2-c8的烷氧基。
最优选的基于有机小分子阴极界面材料具有以下的分子结构式:
本发明的主要优点在于:
1.所发明的有机小分子阴极界面材料具有良好的醇/水溶性,能够溶于甲醇等强极性溶剂,在二氯甲烷中的溶解度有限,因此适合通过正交溶剂法制备器件。
2.所发明的有机小分子阴极界面材料具有很好的阴极修饰能力,可以有效的降低金属阴极的功函数,改善与活性层的欧姆接触,降低接触电阻。
3.所发明的有机小分子阴极界面材料具有较高的导电性,应用于有机太阳能电池中,制备的器件膜厚达到50纳米时,仍然可以获得较高的能量转换效率。
附图说明
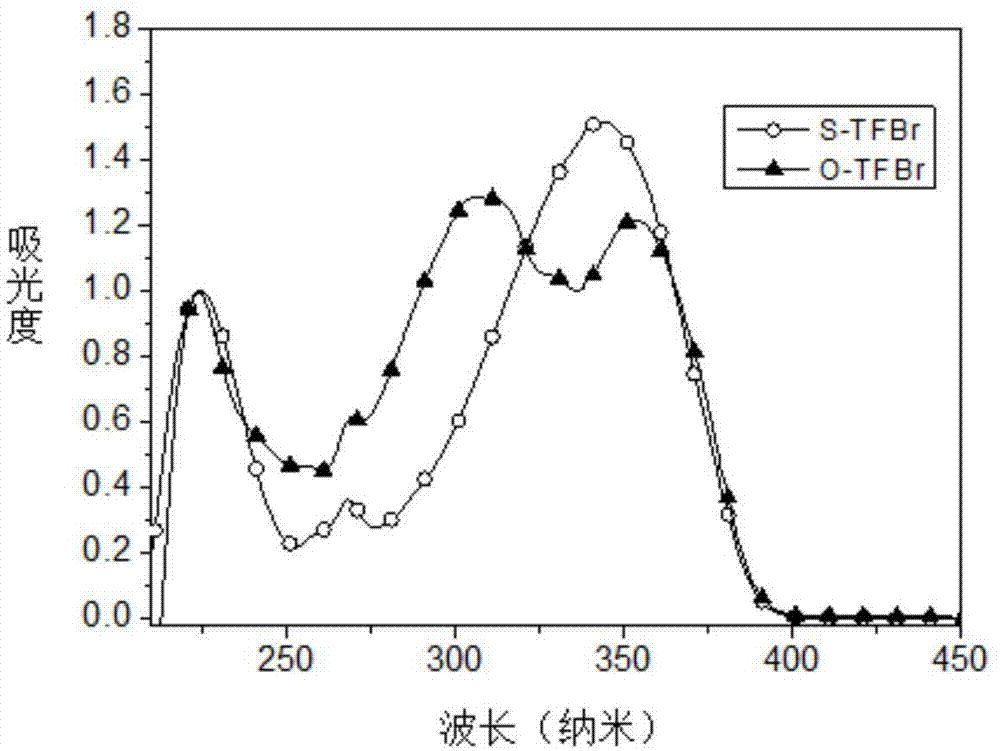
图1为本发明的有机小分子阴极界面材料的紫外-可见光吸收光谱;
图2为本发明的有机小分子阴极界面材料的光电转换效率图;
图3为本发明的有机小分子阴极界面材料o-tfbr在不同膜厚情况下的j-v曲线;
图4为本发明的有机小分子阴极界面材料s-tfbr在不同膜厚情况下的j-v曲线;
图5为应用该类界面材料的有机太阳能电池的结构。
具体实施方式
为使本发明的目的、技术方案和优点更加清楚明白,以下结合具体实施例,并参照附图,对本发明进一步详细说明。
本发明的醇/水溶性有机小分子阴极界面材料的合成路线如下所示:
(i)氢氧化钠(50%),四丁基溴化铵,溴代烷;(ii)2-4,4,5,5-四甲基-[1,3,2]-二氧杂硼烷-9,9-二(3'-溴丙基)芴,四三苯基磷钯,无水碳酸钾,甲苯;(3)四氢呋喃,三甲基胺
实施例1
化合物o-tfbr的制备
具体步骤如下:
(1)化合物1a的合成
将2,7-二溴-9h-芴(0.31克,0.97毫摩尔)和催化量的四丁基溴化铵加入到脱气的二甲基亚砜中(60毫升)。然后加入4毫升氢氧化钠溶液(50%)。此时反应液变成深红色。然后逐滴滴入溶有化合物2-(4-溴甲基苯基)-5-[4-己氧苯基]-[1,3,4]噁二唑(1.0克,2.41毫摩尔)的二甲基亚砜(40毫升)溶液。滴加过程中反应液逐渐变紫。滴加完毕后继续搅拌24小时。反应结束后,将反应液倒入400ml水中,析出固体。过滤,干燥滤饼。然后柱色谱提纯。得到产品0.46克,产率48.0%。
化合物1a的核磁图谱为:1hnmr(400mhz,cdcl3,δ/ppm):7.99-7.94(m,4h,-arh),7.70-7.64(m,1h,arh),7.55-7.47(s,4h,-arh),7.46-7.42(m,1h,arh),7.39-7.36(m,2h,-arh),7.22-7.19(m,2h,-arh),7.01-6.74(m,8h,-arh),4.12-3.99(m,4h,-ch2-),3.49-3.45(m,4h,-ch2-),1.83-1.75(m,4h,-ch2-),1.50-1.31(m,12h,-ch2-),0.94-0.86(m,6h,-ch3).13cnmr(100mhz,cdcl3,δ/ppm):164.4,163.9,161.9,149.0,139.7,138.9,131.0,130.6,128.6,127.6,125.9,122.2,121.5,120.9,116.1,114.9,68.2,57.2,45.3,31.6,29.1,25.7,22.6,14.0.
(2)化合物2a的合成
将化合物1a(0.61克,0.62毫摩尔)、9,9-双(3-溴丙基基)-9h-芴-2-硼酸频哪酯(0.83克,1.56毫摩尔)溶于15毫升脱气甲苯中,然后加入5毫升脱气饱和碳酸钾溶液,氩气保护下加入四三苯基磷钯催化剂。回流反应24小时。反应结束后,加入10毫升水和20毫升二氯甲烷。分离有机层,有机层用无水硫酸镁干燥。过滤,滤液用旋转蒸发器浓缩得粗产品。粗产品柱色谱分离得到淡黄色固体0.92克,产率49%。
化合物2a的核磁图谱为:1hnmr(400mhz,cdcl3,δ/ppm):7.90-7.59(m,22h,arh),7.39-7.36(m,6h,arh),6.99-6.93(s,8h,arh),4.04-3.96(s,4h,-ch2-),3.62-3.55(s,4h,-ch2-),3.19-3.11(m,6h,-ch2-),2.45-2.18(m,6h,-ch2-),1.83-1.75(m,4h,-ch2-),1.56-1.21(m,20h,-ch2-),0.93-0.86(m,6h,-ch3).13cnmr(100mhz,cdcl3,δ/ppm):164.4,164.0,161.9,150.3,150.0,149.6,149.4,148.5,140.9,140.5,140.4,140.3,139.9,139.7,133.8,133.5,131.0,128.6,127.4,127.4,127.3,127.0,126.6,126.4,125.8,123.8,123.4,123.2,122.2,121.8,120.5,120.4,120.2,120.1,119.9,117.9,68.2,56.8,54.3,54.2,45.6,43.5,37.2,34.7,31.5,29.1,27.4,25.7,22.6,14.1.
(3)化合物o-tfbr的合成
在0℃下将化合物2a(0.62克,0.38毫摩尔)溶于20毫升四氢呋喃中,在室温下将三甲胺水溶液(33%,5毫升)逐滴滴加到反应体系中,然后升温至回流,继续反应3小时。反应结束后,用旋转蒸发浓缩反应液至2毫升,然后逐滴滴加到300毫升丙酮中,析出大量固体,过滤得粗产品。粗产品用柱色谱分离得到产物0.34克,产率48.2%。
化合物o-tfbr的核磁图谱为:1hnmr(400mhz,cdcl3,δ/ppm):8.27-8.09(m,4h,arh),8.05-7.83(m,10h,arh),7.65-7.50(m,8h,arh),7.49-7.40(m,6h,arh),7.12-6.90(m,6h,arh),4.10-3.90(s,8h,-ch2-),3.25-3.11(m,4h,-ch2-),2.92-2.84(s,36h,-ch3-br),2.43-2.15(m,8h,-ch2-),1.86-1.65(m,4h,-ch2-),1.56-1.14(m,20h,-ch2-),0.94-0.86(m,6h,-ch3).13cnmr(100mhz,cdcl3,δ/ppm):164.6,164.2,162.4,149.3,148.5,142.7,140.8,140.7,140.2,139.2,131.0,128.4,127.8,127.7,126.8,126.5,125.2,123.4,121.6,120.9,120.5,120.2,114.9,69.2,66.3,58.6,57.5,54.7,54.2,52.0,35.6,31.4,30.8,29.4,28.8,25.4,22.3,18.0,13.0.
实施例2
化合物s-tfbr的制备
具体步骤如下:
(1)化合物1b的合成
将2,7-二溴-9h-芴(0.31g,0.97mmol)和催化量的四丁基溴化铵加入到脱气的二甲基亚砜中(60毫升)。然后加入4毫升氢氧化钠溶液(50%)。此时反应液变成深红色。然后逐滴滴入溶有化合物2-(4-溴甲基苯基)-5-[4-己氧苯基]-[1,3,4]噻二唑(1.0克,2.41毫摩尔)的二甲基亚砜(40毫升)溶液。滴加过程中反应液逐渐变紫。滴加完毕后继续搅拌24小时。反应结束后,将反应液倒入400毫升水中,析出固体。过滤,干燥滤饼。然后柱色谱提纯。得到产品0.46克,产率48.0%。
化合物1b的核磁图谱为:1hnmr(400mhz,cdcl3,δ/ppm):7.98-7.87(m,14h,arh),7.84-7.78(m,2h,arh),7.92-7.89(m,3h,arh),7.76-7.74(m,3h,arh),7.50-7.44(m,10h,arh),7.02-6.96(m,6h,arh),4.04-4.00(t,4h,-ch2-),2.06-2.03(s,4h,-ch2-),1.49-1.46(m,4h,-ch2-),1.37-1.34(m,6h,-ch2-),1.27-1.24(m,6h,-ch2-),0.93-0.91(t,6h,-ch3)13cnmr(100mhz,cdcl3,δ/ppm):167.0,161.6,161.5,149.3,146.5,141.3,139.1,138.9,138.5,138.3,132.7,131.7,130.9,130.8,129.4,128.8,128.4,127.9,127.8,120.1,127.6,126.9,122.5,121.6,120.8,115.0,68.2,65.9,57.2,53.5,46.1,45.1,31.6,29.4,29.1,25.7,22.6,14.1.
(2)化合物2b的合成
将化合物1b(0.63克,0.62毫摩尔)、9,9-双(3-溴丙基基)-9h-芴-2-硼酸频哪酯(0.83克,1.56毫摩尔)溶于15毫升脱气甲苯中,然后加入5毫升脱气饱和碳酸钾溶液,氩气保护下加入四三苯基磷钯催化剂。回流反应24小时。反应结束后,加入10毫升水和20毫升二氯甲烷。分离有机层,有机层用无水硫酸镁干燥。过滤,滤液用旋转蒸发器浓缩得粗产品。粗产品柱色谱分离得到淡黄色固体0.47克,产率46.3%。
化合物2b的核磁图谱为:1hnmr(400mhz,cdcl3,δ/ppm):7.90-7.59(m,22h,arh),7.39-7.36(m,6h,arh),6.97-6.95(s,8h,arh),4.02-3.99(s,4h,-ch2-),3.62-3.58(s,4h,-ch2-),3.18-3.11(m,6h,-ch2-),2.45-2.18(m,6h,-ch2-),1.83-1.75(m,4h,-ch2-),1.55-1.16(m,20h,-ch2-),0.94-0.86(m,6h,-ch3).13cnmr(100mhz,cdcl3,δ/ppm):149.8,149.4,148.7,140.8,140.5,140.4,140.2,139.9,133.6,131.2,129.4,128.3,127.6,127.4,126.9,126.8,126.6,123.3,122.6,121.8,121.4,120.5,120.4,120.2,120.1,117.9,115.0,66.3,56.6,54.3,54.2,45.4,44.9,38.6,34.7,31.5,29.1,27.4,27.2,25.7,22.6,14.1.
(3)化合物s-tfbr的合成
在0℃下将化合物2b(0.47克,0.28毫摩尔)溶于20毫升四氢呋喃中,在室温下将三甲胺水溶液(33%,5毫升)逐滴滴加到反应体系中,然后升温至回流,继续反应3小时。反应结束后,用旋转蒸发浓缩反应液至2毫升,然后逐滴滴加到300毫升丙酮中,析出大量固体,过滤得粗产品。粗产品用柱色谱分离得到产物0.26克,产率49.2%。
化合物s-tfbr的核磁图谱为:1hnmr(400mhz,cdcl3,δ/ppm):8.21-8.09(m,4h,arh),7.85-7.63(m,14h,arh),7.55-7.40(m,10h,arh),7.02-6.90(m,8h,arh),4.00-3.80(s,8h,-ch2-),3.35-3.11(m,4h,-ch2-),2.92-2.84(s,36h,-ch3-br),2.13-2.45(m,8h,-ch2-),1.65-1.86(m,4h,-ch2-),1.14-1.56(m,20h,-ch2-),0.94-0.86(m,6h,-ch3).13cnmr(100mhz,cdcl3,δ/ppm):168.2,167.7,161.9,149.4,148.5,141.8,140.8,140.7,140.2,139.1,131.2,129.1,127.9,127.7,127.4,126.7,126.5,126.2,123.4,121.8,121.5,120.6,120.2,114.9,68.0,66.3,58.8,57.4,54.3,54.2,52.0,35.6,31.4,29.4,28.9,25.5,22.3,18.0,13.1.
实施例3
有机太阳能电池器件的制备
具体步骤如下:
4毫克ptbt与6毫克的pc71bm混合,加入0.5毫升氯苯溶解,得到活性层溶液,备用。通过旋涂的方式在氧化铟锡导电玻璃上制备出一层约40纳米的pedot:pss透明薄膜,在140℃下退火15分钟。然后将退火后的导电玻璃转移至手套箱中,旋涂上述所制备的活性层溶液,然后在将配置好的0.5毫克/升的o-tfbr的阴极界面层的甲醇溶液通过旋涂的方式沉积在活性层之上。最后通过真空蒸镀的方式在阴极界面层上蒸镀一层100纳米厚的金属铝电极。其太阳能电池器件的性能表现为:短路电流为17.64毫安/平方厘米;开路电压为0.78伏特;填充因子为73.4%;在模拟太阳光(a.m.1.5100毫瓦/平方厘米)下的能量转换效率为10.10%。
实施例4
有机太阳能电池器件的制备
具体步骤如下:
4毫克ptbt与6毫克的pc71bm混合,加入0.5毫升氯苯溶解,得到活性层溶液,备用。通过旋涂的方式现在氧化铟锡导电玻璃上制备出一层约40nm的pedot:pss透明薄膜,在140℃下退火15分钟。然后将退火后的导电玻璃转移至手套箱中,旋涂活性层溶液,然后在将配置好的0.5毫克/升的s-tfbr的阴极界面层材料通过旋涂的方式沉积在活性层之上。最后通过真空蒸镀的方式在阴极界面层上蒸镀一层100纳米厚的金属铝电极。其太阳能电池器件的性能表现为:短路电流为17.22毫安/平方厘米;开路电压为0.78伏特;填充因子为70.4%;在模拟太阳光(am1.5;100毫瓦/平方厘米)下的能量转换效率为9.45%。
实施例5
化合物o-tfbr和s-tfbr性能分析如下:
(1)化合物o-tfbr和s-tfbr的光谱分析
化合物o-tfbr和s-tfbr在甲醇溶液和成膜状态下的吸收光谱如图1所示,化合物在200-400纳米范围内有明显吸收,在太阳能电池中并不会与活性层竞争吸收。
(2)化合物o-tfbr和s-tfbr的光伏性能分析
化合物o-tfbr和s-tfbr的光伏性能如图2所示。结果表明基于o-tfbr和s-tfbr的器件都具有良好的光伏性能。基于化合物s-tfbr的器件短路电流为17.22毫安/平方厘米;开路电压为0.78伏特;填充因子为70.4%,在模拟太阳光(am1.5;100毫瓦/平方厘米)下的能量转换效率为9.45%。基于o-tfbr的器件短路电流为17.64毫安/平方厘米;开路电压为0.78伏特;填充因子为73.4%,在模拟太阳光(am1.5;100毫瓦/平方厘米)下的能量转换效率为10.10%。详细的数据参见表1。
表1基于ptb7:pc71bm为活性层,o-tf或s-tf为界面层时的器件参数
不同膜厚情况下的o-tfbr和s-tfbr光伏数据如图3所示,详细的数据参见表2。结果表明通过侧链引入高电子传输性的功能基团,显著增强了器件的电子迁移率,器件的效率对于阴极界面材料的厚度不敏感。
表2阴极界面层在不同膜厚下基于ptb7:pc71bm活性层的器件参数
综上所述,本发明提供了一类侧链含有高电子迁移率的醇/水溶性有机小分子阴极界面材料。这类材料在甲醇中都具有较好的溶解性能,可以通过正交溶剂法制备有机太阳能电池器件。由于引入了高电子传输性能的芳杂环共轭侧链,这类有机小分子界面材料具有较好的导电性,具有膜厚不敏感性,适合于有机太阳能电池的大规模工业制备。以本发明所提供的材料作为阴极界面层时,均获得了超过10%的器件效率。
以上任何实施例的讨论仅为示例性,并不局限于本发明公开的范围(包括权利要求)。为了简明扼要,在本发明的思路下,不同实施例中的技术特征之间的组合以及不同方面的其他变化,并没有在细节中完全提供。因此,凡在本发明的精神和原则之内,所做的任何省略、修改、同等替换、改进等,均包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!