一种利用CRISPRCas9技术敲除大肠杆菌中多粘菌素耐药基因mcr-1的方法与流程
本发明属于微生物基因工程技术领域,具体涉及一种利用crisprcas9技术敲除大肠杆菌中多粘菌素耐药基因mcr-1的方法。
背景技术:
粘菌素是一种古老的抗生素,临床常用的是多粘菌素e和多粘菌素b,它是目前治疗耐碳青霉烯类革兰氏阴性菌的最后储存药物。多粘菌素作为人医临床治疗多重耐药病原菌感染的同时,也在兽医临床广泛用于治疗肠杆菌科菌引发的胃肠道感染,具有重要临床意义。虽然多粘菌素具有较严重的毒副作用,但鉴于革兰氏阴性杆菌多重耐药尤其是碳青霉烯耐药问题严重,2012世卫组织(who)重新将多粘菌素列为人医临床关键药物,成为治疗碳青霉烯类耐药肠杆菌科细菌的最后一道防线。liu等2015年首次在国内发现质粒介导多粘菌素耐药基因mcr-1,证明其耐药机制是通过修饰lps/lpa,减少多粘菌素作用位点,使大肠杆菌对多粘菌素产生耐药性。质粒作为载体介导耐药基因转移,是抗生素耐药性在细菌间传播的主要原因。质粒携带多粘菌素耐药基因mcr-1转移是其水平扩散的重要机制,耐药质粒能在广阔地理区域内的人、动物和环境中及没有明显流行病学关系的病原菌中传播扩散。incx4质粒是携带mcr-1耐药基因洲际传播的主要形式。mcr-1耐药基因以及变体会进一步传播扩散,给人医临床和兽医临床治疗带来严峻挑战,亟需研发新型控制技术阻止mcr-1基因的传播扩散。
crispr/cas系统是一种细菌的免疫防御系统,由细菌在长期抵御外源dna的过程中进化而形成,能够降解入侵的外源dna(病毒或噬菌体等),广泛存在于细菌和古细菌中。crispr/cas系统划分为三种类型:i型、ii型和iii型,其核心蛋白分别是cas3、cas9和cas10。相对于i型和iii型系统需要多种cas蛋白参与反应,并且构成比较复杂的复合体,ii型系统研究最为彻底,只需一个cas9蛋白即能切割目的基因dna。ii型crispr/cas9系统在目前基因编辑方面应用最为广泛。该系统主要由三部分组成:起靶向目的序列的crrna、能够和crrna配对连接的tracrrna以及酶切目的序列的cas9蛋白。crispr序列转录成熟的crrna与tracrrna通过碱基互补配对,形成部分具有双链的rna结构,进而和cas9蛋白形成复合体,剪切外源dna。研究者将crrna和tracrrna整合在同一条单链中,设计出了长度为20bp左右的单链引导rna(singleguiderna,sgrna),sgrna有两个功能,即可以与目的dna序列互补配对,同时引导cas9蛋白,实现基因的敲除等功能。
近年来,crispr/cas9技术已经广泛应用于微生物的基因组编辑和功能研究,尤其在细菌的基因编辑方面,取得了很大的进展。crispr/cas9技术在克服细菌耐药性方面也展现了巨大的潜力,crispr/cas9系统导入细菌后可以切开质粒双链,由于细菌缺失nhej修复机制,被切开的质粒在细菌内被核酸酶裂解。目前已成功应用于β内酰胺类耐药基因blashv-18、blatem、blandm-1等的敲除,使大肠杆菌对抗生素重新敏感。通过查询专利发现数据库中已有一项涉及利用crispr-cas9系统敲除mcr-1的专利:crispr-cas9体外敲除耐药基因mcr-1的方法及其专用细胞穿透肽(申请号:201611123536.8)。但该方法是通过人工构建的mcr-1基因质粒进行的敲除,操作过程复杂、繁琐,无法有效应用于临床菌株。因此,亟需针对广泛携带mcr-1的野生型质粒开展基因敲除,简化crispr-cas9技术流程,为恢复大肠杆菌对多粘菌素敏感性提供快速、简便的技术。
技术实现要素:
有鉴于此,本发明针对上述的问题,提供了一种利用crispr/cas9技术敲除大肠杆菌中多粘菌素耐药基因mcr-1的方法。
为了解决上述技术问题,本发明具体通过以下技术方案实现:
一种利用crisprcas9技术敲除大肠杆菌中多粘菌素耐药基因mcr-1的方法,包括以下步骤:设计如合成sgrna的两条引物序列seqidno.2和seqidno.3,退火形成双链sgrna序列,与psgrna-gfp质粒进行连接,构建psgrna-gfpmcr-1重组质粒,采用电转化法将crispr/cas9敲除系统转化至携带mcr-1的大肠杆菌中,敲除mcr-1基因。
本发明所述的方法中,选择pam区在mcr-1耐药基因第864r位作为靶位点,所述的靶位点的核苷酸序列如seqidno.1所示。
所述的退火反应体系为:sgrnaf(100μm)2μl、sgrnar(100μm)2μl、10×pcrbuffer2μl、ddh2o14μl。所述的退火条件为:95℃10min,关闭水浴锅,自然降温到25℃。
本发明方法中sgrna退火产物与酶切回收的psgrna-gfp质粒连接的反应体系为:退火后的sgrna0.5μl、酶切后的psgrna-gfp质粒1μl、5×t4dnaligasebuffer2μl、t4dnaligase1μl。反应条件为:25℃连接反应2h。
所述的sgrna退火产物与酶切回收的psgrna-gfp质粒的摩尔比为10:1。
本发明方法中酶切反应体系为:psgrna-gfp质粒1.7μl、bamhi限制性内切酶1μl、xbai限制性内切酶1μl、10×nebbuffer5μl、ddh2o42.3μl。酶切条件为:37℃水浴2h,酶切完全后65℃20min灭活内切酶。
本发明的有益效果为:
本发明提供的一种利用crisprcas9技术敲除大肠杆菌中多粘菌素耐药基因mcr-1的方法,通过设计合成sgrna,经与酶切的psgrna-gfp质粒连接,采用电转化法将crispr/cas9敲除系统转化至携带mcr-1的大肠杆菌中,敲除mcr-1基因,使大肠杆菌恢复对多粘菌素敏感,操作简单,简化了crispr-cas9技术流程,为恢复大肠杆菌对多粘菌素敏感性提供快速、简便的技术。
附图说明
图1是靶向mcr-1基因的sgrna位置及周边dna序列;
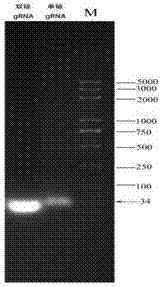
图2是sgrna退火连接产物凝胶电泳图;
图3是psgrna-gfp质粒的双酶切结果。
具体实施方式
下面将结合本发明具体的实施例,对本发明技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
本发明公开了一种利用crispr/cas9技术敲除大肠杆菌中多粘菌素耐药基因mcr-1的方法。采用casot软件筛选mcr-1的靶位点序列,核苷酸序列如seqidno.1所示。根据靶序列,设计合成sgrna序列,包含sgrna-f和sgrna-r,其中sgrna-f核苷酸序列如seqidno.2所示,sgrna-r核苷酸序列如seqidno.3所示。该方法是采用sgrna-f和sgrna-r退火形成双链sgrna序列,与psgrna-gfp质粒进行连接,构建psgrna-gfpmcr-1重组质粒。采用电转化法将crispr/cas9敲除系统(pewtcas9和psgrna-gfpmcr-1质粒)转化至携带mcr-1的大肠杆菌中,敲除mcr-1基因,使大肠杆菌恢复对多粘菌素敏感。
具体过程如下:
1)大肠杆菌中质粒介导多粘菌素耐药基因mcr-1的靶位点筛选及sgrna序列设计
根据sgrna的设计原则,选取casot作为crispr/cas9筛选靶位点的设计软件,设计靶向mcr-1耐药基因的sgrna。在sgrna设计软件中,导入大肠杆菌k12利福平耐药株的全基因组(genbank登录号为cp027060)以及mcr-1耐药基因(genbank登录号为kx570748,locus_tag:pecmcr-1101-0043)后,选择脱靶率较低的位置作为敲除耐药基因mcr-1的靶位点。根据筛选结果,选择pam区在mcr-1耐药基因第864r位作为靶位点。靶向mcr-1基因的sgrna位置及周边dna序列,如图1所示。大肠杆菌中mcr-1的靶位点为cataatacgaatggagtgtgcgg,其中cataatacgaatggagtgtg为靶序列,cgg为pam序列。
合成的sgrna序列,包括sgrna-f和sgrna-r,其中:
sgrna-f:5’-gatcgcataatacgaatggagtgtggttttagag-3’;
sgrna-r:5’-ctagctctaaaaccacactccattcgtattatgc-3’。
2)sgrna序列进行退火连接
对合成好的sgrna序列进行退火连接,全过程在水浴锅中完成,反应体系包括:sgrnaf(100μm)2μl、sgrnar(100μm)2μl、10×pcrbuffer2μl、ddh2o14μl;退火条件为:95℃10min,关闭水浴锅,自然降温到25℃。退火连接的产物于20g/l琼脂糖凝胶中进行电泳,在凝胶成像仪下观察结果,如图2所示。退火连接产物-20℃保存。
3)psgrna-gfpmcr-1重组质粒的构建
①psgrna-gfp质粒双酶切
用bamhi限制性内切酶和xbai限制性内切酶对psgrna-gfp质粒进行双酶切,形成与sgrna两端互补的粘性末端gatc和ctag。psgrna-gfp质粒酶切体系包括:psgrna-gfp质粒1.7μl、bamhi限制性内切酶1μl、xbai限制性内切酶1μl、10×nebbuffer5μl、ddh2o42.3μl;酶切条件为:37℃水浴2h,酶切完全后65℃20min灭活内切酶。酶切反应结束后,取5μl酶切产物,上样于10g/l琼脂糖凝胶进行电泳(原质粒为对照),在凝胶成像仪下观察条带情况,以鉴定酶切是否完全,结果如图3所示。psgrna-gfp酶切质粒的纯化回收按照天根公司的大量琼脂糖凝胶dna回收试剂盒(dp210)的使用说明进行,分光光度计测定其浓度与纯度。
②sgrna退火产物与酶切回收的psgrna-gfp质粒连接
将sgrna退火产物与酶切回收的psgrna-gfp质粒连接按目的基因/酶切质粒载体的摩尔比为10:1的比例进行连接连接反应按照neb公司的t4dnaligase说明书进行,具体反应体系为:退火后的sgrna0.5μl、酶切后的psgrna-gfp质粒1μl、5×t4dnaligasebuffer2μl、t4dnaligase1μl,反应条件为:25℃连接反应2h。
③psgrna-gfpmcr-1重组质粒的鉴定
取含有100μldh5α大肠杆菌感受态细胞的ep管于冰浴中大约15min,加入1μl连接产物,轻轻混匀,冰上静置25min;将感受态细胞转移到42℃水浴热激45s,然后转移至冰浴2min;加入500μllb培养基于细胞中,置于摇床于37℃、200rpm培养1h;吸取100μl培养液均匀涂布到含氨苄青霉素(amp,100μg/ml)lb固体培养基平板上,放于37℃培养箱培养16h。挑取白色的单克隆菌落,在无菌的含有氨苄青霉素(amp,100μ/ml)的lb液体培养基中扩大培养。37℃、200rpm条件下培养过夜。取部分菌液用50%甘油保存在-20℃冰箱中,剩余菌液按照天根生化科技有限公司的质粒小提试剂盒(dp103)提取质粒dna。以m13-f(5’-tgtaaaacgacggccagt-3’)和m13-r(5’-caggaaacagctatgac-3’)为引物进行重组质粒的pcr鉴定。pcr的反应体系为重组质粒载体1μl、m13-f1μl、m13-r1μl、pcrmix22μl;反应条件为:95℃5min;95℃1min、55℃30s、72℃30s,30cycles;72℃10min。pcr反应完成后,取4μl的反应产物进行琼脂糖凝胶电泳检测,以鉴定pcr产物是否正确。对pcr结果为阳性的重组质粒送至成都擎科梓熙生物技术有限公司进行序列测定。
尽管已经示出和描述了本发明的实施例,对于本领域的普通技术人员而言,可以理解在不脱离本发明的原理和精神的情况下可以对这些实例进行多种变化、修改、替换和变型,本发明的范围由所附权利要求及其等同物限定。
序列表
<110>四川大学
<120>一种利用crisprcas9技术敲除大肠杆菌中多粘菌素耐药基因mcr-1的方法
<160>5
<170>siposequencelisting1.0
<210>1
<211>23
<212>dna
<213>mcr-1靶点(mcr-1)
<400>1
cataatacgaatggagtgtgcgg23
<210>2
<211>34
<212>dna
<213>人工序列(artificialsequence)
<400>2
gatcgcataatacgaatggagtgtggttttagag34
<210>3
<211>34
<212>dna
<213>人工序列(artificialsequence)
<400>3
ctagctctaaaaccacactccattcgtattatgc34
<210>4
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>4
tgtaaaacgacggccagt18
<210>5
<211>17
<212>dna
<213>人工序列(artificialsequence)
<400>5
caggaaacagctatgac17
- 还没有人留言评论。精彩留言会获得点赞!