具有改良光学的乙烯类聚合物的制作方法
本申请要求2017年3月21日提交的美国临时申请62/474134的权益。
背景技术:
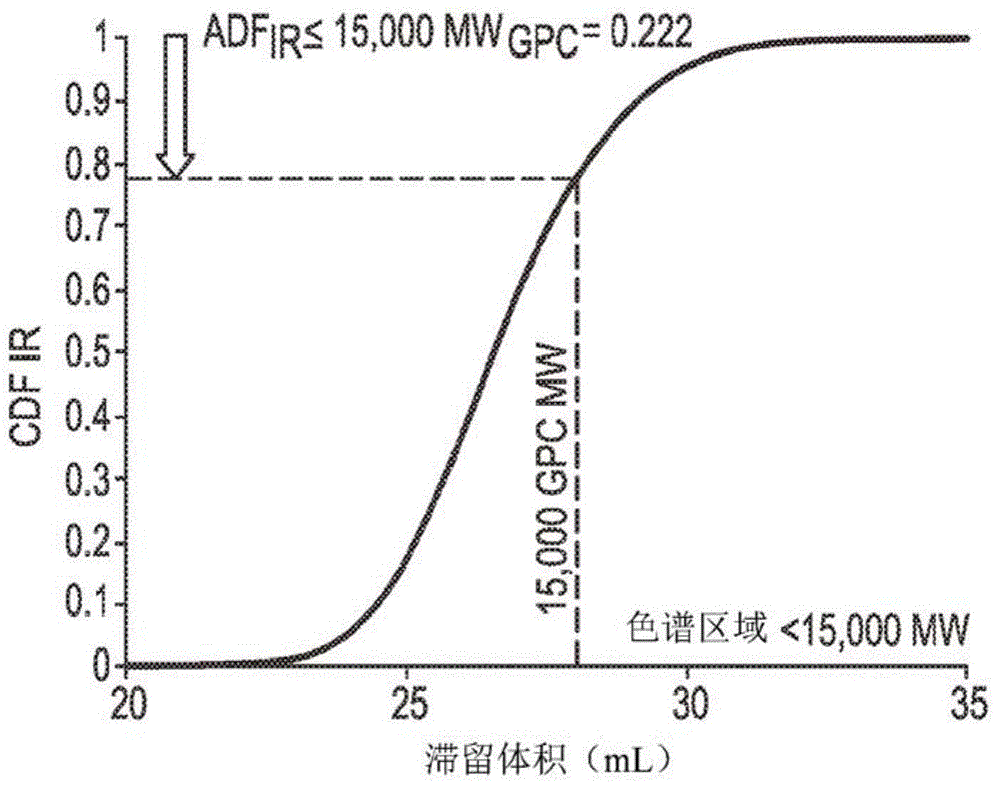
:需要具有良好光学和良好可处理性且可用于吹制膜和收缩膜应用的ldpe树脂。ldpe树脂描述于以下专利文献中:美国专利8415442、美国专利9243087、美国专利9068032、美国专利9228036(还参见美国公开案2014/0316094)、美国专利8822601、美国专利8871887、美国专利8916667(还参见美国专利9303107)、美国公开案2016/0137822、美国公开案2016/0083568、wo2017/146981和wo2017/201110。然而,仍需要具有良好光学和可处理性且可用于单层或多层吹制膜或收缩膜的ldpe树脂。这些需求已通过以下发明满足。技术实现要素:一种包括乙烯类聚合物的组合物,其中所述乙烯类聚合物包括以下特性:a)mw(abs)为130,000至162,00g/mol;b)熔融指数(i2)为1.5至3.0g/10min;c)对于分子量≥500,000g/mol,adfls为0.350至0.450。附图说明图1描绘对于实例1的cdfir测定的“累积ir面积相对于滞留体积”。图2描绘对于实例1的cdfls测定的“累积ls面积相对于滞留体积”。图3描绘对于实例1的cdfdv测定的“累积dv(粘度计)面积相对于滞留体积”。图4描绘用以生产乙烯类聚合物(ldpe)实例的聚合系统的方块图。图5描绘对于本发明实例和比较实例,熔融强度作为熔融指数的函数。具体实施方式一种包括乙烯类聚合物的组合物,其中所述乙烯类聚合物包括以下特性:a)mw(abs)为130,000至162,00g/mol;b)熔融指数(i2)为1.5至3.0g/10min;和c)对于分子量≥500,000g/mol,adfls为0.350至0.450。组合物可包含两个或多于两个如本文所述的实施例的组合。乙烯类聚合物可包含两个或多于两个如本文所述的实施例的组合。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物为低密度聚乙烯(ldpe)。在本文所述的一个实施例或实施例的组合中,对于分子量≤15,000g/mol,乙烯类聚合物进一步包括0.200至0.250的adfir。在本文所述的一个实施例或实施例的组合中,对于分子量≥200,000g/mol,乙烯类聚合物进一步包括0.180至0.240的adfdv。在本文所述的一个实施例或实施例的组合中,聚合物进一步包括1.8至2.0的mw(abs)/mw(conv)。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物进一步包括0.820dl/g至0.880dl/g的iv(abs)。在本文所述的一个实施例或实施例的组合中,对于分子量≥200,000g/mol,乙烯类聚合物进一步包括≤0.210的adfdv。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物进一步包括gpcbr≤1.65、或≤1.60、或≤1.58、或≤1.56。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物的密度为0.923至0.927g/cc、或0.923至0.926g/cc、或0.924至0.926g/cc(1cc=1cm3)。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物的“45°光泽”为大于72%,且2%md正割模数大于30,000psi。在一个实施例或本文所述的实施例的组合中,乙烯类聚合物的md标准化撕裂比cd标准化撕裂的比率为小于1.1且大于0.8。在一个实施例或本文所述的实施例的组合中,乙烯类聚合物的md收缩张力比cd收缩张力的比率为小于7.0,其中cd收缩张力大于0.9psi。在各实施例中,膜厚度为2.0±0.3密耳。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物的md收缩张力比cd收缩张力的比率≤8.0,或≤7.0,或≤6.0,或≤5.0。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物的md比cd收缩张力的比率为≥1.0,或≥2.0,或≥3.0,或≥4.0。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物的md收缩张力≤10.0psi,或≤8.0psi,或≤6.0psi,或≤5.0psi。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物的md收缩张力≥1.0psi,或≥2.0psi,或≥3.0psi,或≥4.0psi。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物的cd收缩张力≤2.0psi,或≤1.75psi,或≤1.5psi,或≤1.25psi。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物的cd收缩张力≥0.5psi,或≥0.75psi,或≥0.9psi,或≥1.0psi。在各实施例中,膜厚度为2.0±0.3密耳。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物的adfir或adfir(在分子量(mw)≤15,000g/mol下)≥0.19,或≥0.20,或≥0.21,或≥0.22。在另一实施例中,乙烯类聚合物为ldpe。ldpe为本领域中已知,且是指使用自由基高压(≥100mpa(例如100-400mpa))聚合制备的乙烯均聚物。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物的adfir(在分子量(mw)≤15,000g/mol下)≤0.25,或≤0.24,或≤0.23。在另一实施例中乙烯类聚合物为ldpe。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物的adfls或adfls(在分子量(mw)≥500,000g/mol下)≥0.355,或≥0.360,或≥0.365。在另一实施例中,乙烯类聚合物为ldpe。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物的adfls(在分子量(mw)≥500,000g/mol下)≤0.400,或≤0.395,或≤0.390。在另一实施例中,乙烯类聚合物为ldpe。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物的adfdv或adfdv(在分子量(mw)≥200,000g/mol下)≥0.165,或≥0.170,或≥0.175,或≥0.180。在另一实施例中,乙烯类聚合物为ldpe。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物的adfdv(在分子量(mw)≥200,000g/mol下)≤0.200,或≤0.195,或≤0.190。在另一实施例中乙烯类聚合物为ldpe。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物的mw(abs)为130,000至160,000g/mol,或自135,000至160,000g/mol,或自140,000至160,000g/mol。在另一实施例中,乙烯类聚合物为ldpe。在本文所述的一个实施例或实施例的组合中,聚合物的mz(abs)为2,000,000至5,000,000g/mol,或2,500,000至4,500,000g/mol,和/或3,000,000至4,000,000g/mol。在另一实施例中,乙烯类聚合物为ldpe。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物的mz(abs)/mw(abs)为12.0至30.0,或15.0至25.0,或18.0至23.0。在另一实施例中,聚合物为ldpe。在一个实施例中,乙烯类聚合物的mw(abs)/mw(conv)比率≥1.70,或≥1.75,或≥1.80,或≥1.85。在另一实施例中,聚合物为ldpe。在一个实施例中,乙烯类聚合物的mw(abs)/mw(conv)比率为1.70至3.00,或1.70至2.50,或1.70至2.20,或1.70至2.00。在另一实施例中,聚合物为ldpe。在本文所述的一个实施例或实施例的组合中,聚合物的gpcmw(conv)为60,000至200,000g/mol,或65,000至150,000g/mol,或70,000至130,000g/mol,或75,000至100,000g/mol。在另一实施例中,乙烯类聚合物为ldpe。在本文所述的一个实施例或实施例的组合中,聚合物的mw(conv)/mn(conv)≥4.6或≥4.8或≥5.0。在另一实施例中,聚合物为ldpe。在本文所述的一个实施例或实施例的组合中,聚合物的mw(conv)/mn(onv)≤6.0或≤5.8或≤5.6。在另一实施例中,乙烯类聚合物为ldpe。在本文所述的一个实施例或实施例的组合中,聚合物的mn(conv)为10,000至20,000g/mol,或12,500至18,500g/mol,或14,000至17,000g/mol,或15,000至16,000g/mol。在另一实施例中,聚合物为ldpe。在一个实施例中,乙烯类聚合物的z-平均分子量mz(conv)≥200,000g/mol、或≥230,000g/mol或≥250,000g/mol。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物的z-平均分子量mz(conv)≤400,000g/mol、或≤350,000g/mol或≤320,000g/mol。此外,乙烯类聚合物为ldpe。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物的gpcbr值为1.2至3.0,或1.3至2.5,或1.4至2.0,或1.4至1.8。在另一实施例中,乙烯类聚合物为ldpe。在一个实施例中,乙烯类聚合物的lcbf值为1.0至3.0,或1.1至2.5,或1.2至2.0,或1.2至1.7。在另一实施例中,乙烯类聚合物为ldpe。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物的固有粘度(通过利用gpc的在线粘度计绝对)或iv(主体)>0.750或>0.800。在本文所述的一个实施例或实施例的组合中,聚合物的固有粘度(通过在线利用gpc的粘度计绝对)或iv(abs)≤0.920或≤0.900。在另一实施例中,乙烯类聚合物为ldpe。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物在190℃下的熔融强度<-1.87×(在190℃下的i2)+10cn。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物在190℃下的熔融强度≥-1.87×(在190℃下的i2)+6cn。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物在190℃下的熔融强度≥-1.87×(在190℃下的i2)+8cn。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物在190℃下的熔融强度<-1.87×(在190℃下的i2)+10cn,且熔融强度≥-1.87×(在190℃下的i2)+8cn。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物在190℃下的熔融强度<-1.87×(在190℃下的i2)+10cn且≥-1.87×(在190℃下的i2)+6cn。对于以上各等式,“1.87系数”的单位如下:“(cn)/(g/10min)”。在另一实施例中,乙烯类聚合物为ldpe。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物在0.1rad/s和190℃下的熔融粘度≥2,000pa·s,或≥3,000pa·s,或≥4,000pa·s,或≥4,500pa·s。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物在0.1rad/s和190℃下的熔融粘度≤10,000pa·s,或≤9,000pa·s,或≤8,500pa·s,或≤8,000pa·s,或≤7,500pa·s,或≤7,000pa·s。在另一实施例中,乙烯类聚合物为ldpe。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物的熔融粘度比(v0.1/v100)在190℃下≥5.0,或≥7.5,或≥10。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物的粘度比(v0.1/v100,在190℃下)为5至20,或7.5至15,或8至14,或9至13。在另一实施例中,乙烯类聚合物为ldpe。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物的正切δ(在190℃下以0.1rad/s)≤20,或≤10,或≤8.0,或≤7.0。在一个实施例中,乙烯类聚合物的正切δ(在190℃下以0.1rad/s)为2.0至15,或3.0至13,或4.0至10,或5.0至7.0。在另一实施例中,乙烯类聚合物为ldpe。在一个实施例中,乙烯类聚合物的熔融强度(ms)>3cn或>4cn或>5cn。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物的熔融指数(i2)为1.6至3.0g/10min,或1.8至3.0g/10min,或2.0至2.9g/10min。在另一实施例中,乙烯类聚合物为ldpe。在一个实施例中,乙烯类聚合物的密度为0.910至0.940g/cc,或0.910至0.930g/cc,或0.915至0.9275g/cc,或0.920至0.9275g/cc,或0.923至0.927g/cc(1cc=1cm3)。在另一实施例中,乙烯类聚合物为ldpe。在本文所述的一个实施例或实施例的组合中,按聚合物的重量计,乙烯类聚合物的%己烷可萃取物为0.5至4.0wt%,或0.5至3.0wt%,或0.5至2.0wt%,或0.75至1.25wt%。在另一实施例中,乙烯类聚合物为ldpe。在本文所述的一个实施例或实施例的组合中,如通过13cnmr所测定,乙烯类聚合物每1000个总碳原子有≥0.1戊基(c5)。在本文所述的一个实施例或实施例的组合中,如通过13cnmr所测定,乙烯类聚合物每1000个总碳原子有≥0.2戊基(c5)基团(分支),或每1000个总碳原子有≥0.5戊基,或每1000个总碳原子有≥1戊基,或每1000个总碳原子有≥1.2戊基,或每1000个总碳原子有≥1.5戊基,其中戊基相当于c5基团。在另一实施例中,聚合物为ldpe。在本文所述的一个实施例或实施例的组合中,如通过13cnmr所测定,聚合物每1000个总碳原子有<0.1c1(甲基)。在另一实施例中,聚合物为ldpe。在一个实施例中,如通过13cnmr所测定,乙烯类聚合物具有每1000个总碳原子1.0至5.0的1,3二乙基分支,或每1000个总碳原子1.5至4.0的1,3二乙基分支,或每1000个总碳原子1.75至3.5的1,3二乙基分支。在另一实施例中,乙烯类聚合物为ldpe。在本文所述的一个实施例或实施例的组合中,如通过13cnmr所测定,乙烯类聚合物具有每1000个总碳原子1.0至5.0的c6+分支,或每1000个总碳原子2.0至4.0的c6+分支,或每1000个总碳原子2.0至3.0的c6+分支。在另一实施例中,聚合物为ldpe。在一个实施例中,如通过13cnmr所测定,乙烯类聚合物具有每1000个总碳原子0.5至3.0个来自丁烯的隔离c2,或每1000个总碳原子0.75至2.0个来自丁烯的隔离c2,或每1000个总碳原子1.0至2.0个来自丁烯的隔离c2。在另一实施例中,聚合物为ldpe。在本文所述的一个实施例或实施例的组合中,如通过13cnmr所测定,乙烯类聚合物具有每1000个总碳原子0.25至2.0个在第四碳原子上的c2,或每1000个总碳原子0.5至1.5个在第四碳原子上的c2,或每1000个总碳原子0.6至1.2个在第四碳原子上的c2。在另一实施例中,聚合物为ldpe。在一个实施例中,如通过1hnmr所测定,乙烯类聚合物具有每1000个总碳原子0.05至0.15个乙烯基,或每1000个总碳原子0.075至0.13个乙烯基,或每1000个总碳原子0.09至0.12个乙烯基。在另一实施例中,聚合物为ldpe。在一个实施例中,如通过1hnmr所测定,乙烯类聚合物具有每1000个总碳原子0.1至0.3个顺式和反式基团(亚乙烯基),或每1000个总碳原子0.15至0.25个顺式和反式。在另一实施例中,聚合物为ldpe。在一个实施例中,如通过1hnmr所测定,乙烯类聚合物具有每1000个总碳原子0.05至0.2个亚乙烯基,或每1000个总碳原子0.075至0.15个亚乙烯基。在另一实施例中,聚合物为ldpe。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物的结晶温度为90.0℃至110.0℃、或95.0℃105.0℃、或100.0℃至101.0℃。在另一实施例中,乙烯类聚合物为ldpe。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物的熔融温度为95.0℃至115.0℃、或97.0℃至114.0℃、或110.0℃至114.0℃。在另一实施例中,乙烯类聚合物为ldpe。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物在高压(p大于100mpa)自由基聚合工艺中形成。在另一实施例中,聚合物为ldpe。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物以按组合物的重量计≥10wt%的量存在。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物以按组合物的重量计10至90wt%、或20至40wt%的量存在。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物以按组合物的重量计60至90wt%、或65至85wt%的量存在。在另一实施例中,聚合物为ldpe。在一个实施例中,乙烯类聚合物以按组合物的重量计1至10wt%、或1.5至8wt%、或3至6wt%的量存在。在一个实施例中,乙烯类聚合物以按组合物的重量计80至100wt%、或85至97wt%、或85至95wt%的量存在。在另一实施例中,聚合物为ldpe。在本文所述的一个实施例或实施例的组合中,组合物进一步包括另一种乙烯类聚合物。适合的其它乙烯类聚合物包含但不限于:dowlex聚乙烯树脂、tuflin线性低密度聚乙烯树脂、elite或eliteat增强聚乙烯树脂或innate精密包装树脂(均可自陶氏化学公司(thedowchemicalcompany)获得)、高密度聚乙烯(d≥0.96g/cc)、中密度聚乙烯(密度为0.935至0.955g/cc)、exceed聚合物和enable聚合物(两者来自埃克森美孚(exxonmobil))、ldpe和eva(乙烯乙酸乙烯酯)。在一个实施例中,第二乙烯类聚合物为乙烯/c3-c8α烯烃共聚物,如lldpe(线性低密度聚乙烯)。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物以按乙烯类聚合物和第二乙烯类聚合物的总重量计10wt%至90wt%的量存在。在本文所述的一个实施例或实施例的组合中,乙烯类聚合物以按乙烯类聚合物和第二乙烯类聚合物的总重量计3wt%至97wt%的量存在。在本文所述的一个实施例或实施例的组合中,组合物进一步包括丙烯类聚合物。适合的丙烯类聚合物包含聚丙烯均聚物、丙烯/α烯烃互聚物和丙烯/乙烯互聚物。聚合物包含冲击改性聚丙烯、全同立构聚丙烯、非规聚丙烯和无规指定versify塑性体和弹性体(陶氏化学公司)和vistamaxx(埃克森美孚化学公司(exxonmobilchemicalco.))。在本文所述的一个实施例或实施例的组合中,组合物进一步包括非均相支链乙烯/α-烯烃互聚物或共聚物。在另一实施例中,此类非均相支链互聚物或共聚物的密度为0.89至0.94g/cc,或0.90至0.93g/cc。在一个实施例中,组合物包括按组合物的重量计小于5ppm、或小于2ppm、或小于1ppm、或小于0.5ppm硫。本发明还提供一种制品,其包含至少一种由本发明组合物形成的组分。在另一实施例中,制品为膜。在另一实施例中,制品为涂层。本发明还提供用于形成前述实施例中任一项的乙烯类聚合物的工艺,所述工艺包括使包括乙烯的混合物在至少一种管状反应器中聚合。本发明还提供用于形成此类乙烯类聚合物的工艺,所述工艺包括使包括乙烯的混合物在至少一个管状反应器和至少一个高压釜反应器的组合中聚合。本发明组合物可包括如本文所述的两个或多于两个实施例的组合。本发明乙烯类聚合物可包括如本文所述的两个或多于两个实施例的组合。本发明ldpe可包括如本文所述的两个或多于两个实施例的组合。本发明制品可包括如本文所述的两个或多于两个实施例的组合。本发明膜可包括如本文所述的两个或多于两个实施例的组合。本发明方法可包括如本文所述的两个或多于两个实施例的组合。方法为了制造本发明乙烯类聚合物(包含本发明ldpe),通常使用高压、自由基引发的聚合方法。通常,将具有一个或多个反应区的带夹套的管用作反应器。适合但非限制性的反应器长度可为100至3000米(m),或1000至2000米。反应器的反应区起点通常由侧面注射反应的引发剂(乙烯、链转移剂(或短链聚合物)以及其任何组合)来界定。高压方法可在一个或多个具有一个或多个反应区的管状反应器中进行,或在各自包括一个或多个反应区的高压釜和管状反应器的组合中进行。链转移剂可用以控制分子量。在一个优选实施例中,将一种或多种链转移剂(cta)添加至本发明方法。可使用的典型cta包含但不限于丙烯、正丁烷、1-丁烯、异丁烷、丙醛、和甲基乙基酮。在一个实施例中,用于所述方法中的cta的量为总反应混合物的0.03至10重量%。用于制造乙烯类聚合物的乙烯可为经纯化乙烯,其可通过自回路再循环流去除极性组分或通过使用反应系统配置获得,使得仅新鲜乙烯用于制造本发明聚合物。仅需要经纯化乙烯以制造乙烯类聚合物不为典型的。在此类情况下,可使用来自再循环回路的乙烯。在一个实施例中,乙烯类聚合物为ldpe。添加剂和应用本发明组合物可包含一种或多种添加剂。添加剂包含但不限于稳定剂、塑化剂、抗静电剂、颜料、染料、成核剂、填充剂、增滑剂、阻燃剂、加工助剂、烟雾抑制剂、粘度控制剂和抗阻塞剂。组合物可例如包括以组合物的重量计小于10(组合重量)%的一种或多种添加剂。在一个实施例中,本发明的聚合物经一种或多种稳定剂处理,所述稳定剂例如抗氧化剂,如irganox1010、irganox1076和irgafos168(汽巴精化(cibaspecialtychemicals))。可在多种热塑性制造过程中采用本发明的聚合物以制造有用的对象,包含但不限于单层和多层膜;模制品,如吹塑模制、注射模制或旋转模制对象;涂层(例如挤压涂层);纤维;和织物或非织物。聚合物本发明可用于多种膜,包括但不限于食品包装、消费品、工业、农业(应用或膜)、层合膜、新鲜切割生产品膜、肉类膜、奶酪膜、糖果膜、透明收缩膜、整理收缩膜、拉伸膜、青贮料膜、温室膜、熏蒸膜、衬膜、拉伸罩、重负载装运袋、宠物食品、夹层袋、密封层和尿布背层。本发明聚合物可用于导线和线缆涂布操作,在片材挤压中用于真空成形操作,和形成模制品,包含使用注射模制、吹塑模制或旋转模制方法。定义如本文所用,术语“聚合物”是指通过聚合相同或不同类型的单体制备的聚合化合物。通用术语聚合物由此涵盖术语均聚物(用以指代由仅一种类型的单体制备的聚合物),其中应理解低量杂质(例如低量(例如≤1.0wt%,或≤0.5wt%,或≤0.3wt%)的cta)可并入至聚合物结构中;和如下文所定义的术语互聚物。杂质可并入至聚合物中和/或在聚合物内。如本文所用的术语“互聚物”是指通过使至少两种不同类型的单体聚合尔制备的聚合物。通用术语互聚物包含共聚物(用以指代由两种不同类型的单体制备的聚合物)和由多于两种不同类型的单体制备的聚合物。如本文所用,术语“乙烯类聚合物”是指包含50wt%或大量聚合乙烯单体(以聚合物的重量计)且任选地可含有至少一种共聚单体的聚合物。如本文所用,术语“丙烯类聚合物”是指包含大部分量的聚合丙烯单体(以聚合物的重量计)且任选地可包含至少一种共聚单体的聚合物。如本文中所使用,术语“组合物”包括:包含所述组合物的材料的混合物,以及由所述组合物的材料形成的反应产物和分解产物。如所使用,术语“掺合物”或“聚合物掺合物”是指两种或多于两种聚合物的混合物。一种掺合物可为可混溶,或可不为可混溶的(并非在分子水平上分离)。种掺合物可为相分离,或可不为相分离的。一种掺合物可含有或可不含有一个或多个域构形,如根据传输电子波谱学、光散射、x射线散射和其它本领域中已知的方法所测定的那般。所述掺合物可由使两个或更多个聚合物在宏观水平上(例如熔融掺和树脂或混配)或微观水平上(例如同时形成于相同反应器内)实体地混合来实现。术语“包含”、“包括”、“具有”和其衍生词并不打算排除任何额外组分、步骤或程序的存在,无论其是否具体地公开。为避免任何疑问,除非相反陈述,否则经由使用术语“包含”所主张的所有组成物均可包括任何额外添加剂、佐剂或化合物,无论聚合或以其它方式。术语“基本上由...组成”自任何随后列举范围中排除任何其它组分、步骤或程序,对于可操作性而言并非必需的那些组分、步骤或程序除外。术语“由...组成”排除未具体叙述或列举的任何组分、步骤或程序。测试方法(使用目前版本的astm测试方法)密度根据astmd4703附录a1程序c制备用于密度测量的样品。将约7g样品置于“2″×2″×135密耳厚度”模具中,且此在374°f(190℃)下以3,000lbf按压6分钟。将压强增加至30,000lbf持续4分钟。此随后在30,000lbf下以15℃/分钟冷却至约40℃的温度。随后自模具移出“2″×2″×135密耳”聚合物样品(薄板),且用1/2″×1″模切机(diecutter)由薄板切割出3个样品。测量在样品按压1小时内使用astmd792方法b进行,且报告3次测量的平均值。熔融指数熔融指数(mi)或i2根据astmd1238,条件190℃/2.16kg程序b测量,且以每10分钟洗脱克数(g/10min)为单位报告。己烷可萃取物聚合物团粒(来自聚合粒化方法,不经其它改变;每一“1英寸×1英寸”正方形膜约2.2克)以厚度为3.0-4.0密耳的卡弗按压(carverpress)按压。将团粒在190℃下以40,000lbf按压3分钟。穿戴无残留手套(pip*cleanteam*cottonlisle检测手套,部件号:97-501)以防止来自操作员手部的残留油对膜的污染。将各膜修整成“1英寸×1英寸”正方形,且称重(2.5±0.05g)。膜在含有约1000ml己烷的己烷容器中于49.5±0.5℃下在热水浴中萃取2小时。己烷为异构“己烷”混合物(例如己烷(最佳值)、费舍尔(fisher)化学物质、用于hplc的高纯度移动相和/或用于gc应用的萃取溶剂)。在两小时后将膜去除,在纯净的己烷中冲洗,且在完全真空(isotemp真空烘箱,型号281a,处于约30英寸汞)下于真空烘箱中干燥(80±5℃)两小时。随后将膜置于干燥器中,且使其冷却至室温维持一小时的一分钟。随后将膜再称重,且计算由于在己烷中萃取的质量损失的量。此方法是基于21crf177.1520(d)(3)(ii),其中与fda方案的一个差别为使用己烷代替正己烷;报告3次测量的平均值。核磁共振(13cnmr)通过在10mmnmr管中将约“3g含有0.025mcr(acac)3的四氯-乙烷-d2/邻二氯苯的50/50混合物”添加至“0.25g聚合物样品”制备各样品。随后使用加热块和热风枪通过将管和其内含物加热至150℃来使样品溶解且均匀化。目测检查各经溶解样品以确保均匀性。所有数据使用配备有bruker双dul高温cryoprobe的bruker400mhz光谱仪收集。使用六秒脉冲重复延迟、90度翻转角和非门控去耦(inversegateddecoupling)和120℃的样品温度获取数据。全部测量在锁定模式下在不旋转样品上进行。在30.0ppm下将13cnmr化学位移内标为eee三元组(triad)。c6+值为ldpe中c6+分支的直接测量,其中长分支未区别于链末端。代表自所有链末端或六个或多于六个碳原子的分支末端的第三个碳的32.2ppm峰值用以测定c6+值。使用测定聚合物组合物的熟知nmr光谱方法测定lldpe中的c6分支。astmd5017-96,j.c.randall等人于“nmr与大分子(nmrandmacromolecules)”acs研讨会系列(acssymposiumseries)247,j.c.randall,编,《美国化学学会(am.chem.soc.)》,华盛顿(washington,d.c.),1984,第9章;和j.c.randall于“聚合物序列测定(polymersequencedetermination)”,学术出版社(academicpress),纽约(newyork)(1977)中提供通过nmr光谱法的聚合物分析的普遍方法。其它所关注的峰值列出于表a中。表a核磁共振(1hnmr)通过在norell1001-7、10mmnmr管中将约130mg样品添加至具有0.001mcr(acac)3的“3.25g50/50重量%的四氯乙烷-d2/过氯乙烯”制备各样品。经由插入至管中的移液管通过鼓泡n2通过溶剂来冲洗样品,持续约5分钟以防止氧化。管经封盖,用teflon带密封,且随后在室温下浸泡过夜以促进样品溶解。在115℃下加热样品且涡旋以保证均匀性。在配备有bruker双dul高温cryoprobe的brukeravance400mhz光谱仪上进行1hnmr,且样品温度为120℃。执行两个实验以获得光谱,对照光谱用于定量总聚合物质子,以及双预饱和实验,其抑制强烈聚合物主结构主链峰,且实现用于定量端基的高灵敏度光谱。用zg脉冲、16扫描、aq1.64s、d114s执行对照。用改良脉冲序列、100扫描、aq1.64s、预饱和延滞1s、弛豫延滞13s执行双预饱和实验。将来自tce-d2(在6.0ppm下)中的残余1h的信号积分,且设定为值100,且自3至-0.5ppm的积分用作来自对照实验中的全部聚合物的信号。对于预饱和实验,tce信号也设定为100,且获得对于不饱和度(亚乙烯基(顺式和反式)在约5.40ppm至5.60ppm,三取代在约5.16ppm至5.35ppm,乙烯基在约4.95ppm至5.15ppm,且亚乙烯基在约4.70ppm至4.90ppm)的相对应积分。熔融强度熔融强度测量在连接至gottfertrheotester2000毛细管流变仪的gottfertrheotens71.97(inc.;rockhill,sc)上进行。用配备有长度为30mm、直径为2.0mm、和纵横比(长度/直径)为15的平面进入角(180度)的rheotester2000毛细管流变仪馈入熔融样品(约25至30克)在使样品在190℃下平衡10分钟之后,活塞以0.265毫米/秒的恒定活塞速度运行。标准测试温度为190℃。将样品以2.4mm/sec2的加速度单向地拉伸至位于模具下方100mm的一组加速夹。拉力记录为夹持辊的起始速度的函数。熔融强度报导为平均平线区力(cn)且断裂时的速度以mm/s为单位报告。以下条件用于熔融强度测量中:推杆速度=0.265mm/sec;轮加速度=2.4mm/sec2;毛细管直径=2.0mm;毛细管长度=30mm;且筒直径=12mm。动态机械光谱学(dms)将树脂在350°f下以20,000lbf在空气中维持6.5分钟压缩模制成“3mm厚度×1英寸”环形薄板。随后自压机取出样品且置放于柜台上以冷却。在氮气净化下使用配备有25mm(直径)平行板的tainstruments“先进流变膨胀系统(advancedrheometricexpansionsystem;ares)”进行恒定温度频率清扫。样品置放于板上,且使其在190℃下熔融5分钟。随后将板闭合至“2mm”的间隙,修整样品(去除扩展超出“25mm直径”板的外周的额外样品),且随后开始测试。方法置入有另外五分钟延迟,以允许温度平衡。在190℃下以0.1至100rad/s的频率范围进行实验。应变幅度恒定在10%。测量复数粘度η*、正切(δ)或正切δ、0.1rad/s下的粘度(v0.1)、100rad/s下的粘度(v100)和粘度比(v0.1/v100)。三重检测器凝胶渗透色谱法(tdgpc)色谱系统由以下组成:配备有内部ir5红外检测器(ir5)的polymerchargpc-ir(西班牙巴伦西亚市(valencia,spain))高温gpc色谱仪连接至精确度检测器(nowagilenttechnologies)2-角度雷射光散射(ls)检测器型号2040,且接着polymerchar4-毛细管粘度检测器(三个检测器串联)。对于所有光散射测量,将15度角用于测量目的。自动进样器烘箱隔室设定在160摄氏度且管柱隔室设定在150摄氏度。所使用的柱为四个各30cm的agilent“混合a”柱,且各自通过20微米线性混合床颗粒填充。所使用的色谱溶剂为1,2,4-三氯苯,其含有200ppm的丁基化羟基甲苯(bht)。溶剂源经氮气鼓泡。注入体积为200微升且流动速率为1.0ml/min。用至少20窄分子量分布聚苯乙烯标准物进行gpc柱设置的校准,所述聚苯乙烯标准物的分子量在580至8,400,000g/mol范围内。这些标准物配置于6种“调配”混合物中,其中独立分子量之间的间隔为约十倍。标准品购自agilenttechnologies。对于分子量≥1,000,000g/mol,聚苯乙烯标准物以“于50毫升溶剂中0.025克”制备,且对于分子量<1,000,000g/mol以“于50毫升溶剂中0.05克”制备。聚苯乙烯(ps)标准物在80℃下在轻微搅拌下溶解30分钟。使用等式1(如在williams和ward,j.《聚合物科学(polym.sci.)》,《聚合物快报(polym.let.)》,6,621(1968)中所描述)将ps标准物峰分子量(ir5检测器)转化为聚乙烯分子量:m聚乙烯=a×(m聚苯乙烯)b(等式1)其中m为分子量,a的值为0.4315且b等于1.0。使用第五阶多顶式以拟合对应聚乙烯当量校准点。对a进行小调整(约0.415至0.44)以修正柱分辨率和带变宽效果,使得在52,000g/mol(mw)下获得nist标准nbs1475。gpc柱设置的总平板计数用eicosane(以0.04g于50毫升tcb(1,2,4-三氯-苯)稳定溶剂中制备,且在缓慢搅拌下溶解20分钟)来进行。在200微升注入上根据以下等式测量板计数(等式2)和对称性(等式3):其中rv为以毫升为单位的滞留体积,峰宽以毫升为单位,最大峰值为峰的最大高度,且1/2高度为最大峰值的1/2高度:其中rv为以毫升为单位的滞留体积,且峰宽以毫升为单位,“最大峰值”为对应于色谱图“rv位置”的最大ir信号高度,“十分之一高度”最大峰值的1/10高度,其中“背峰”是指比最大峰值较晚的处于信号滞留体积的峰值尾部(处于最大峰值1/10高度),且其中“前峰”是指比最大峰值较早的处于信号滞留体积的峰值前部(处于最大峰值1/10高度)。色谱系统的板计数应大于24,000且对称性应介于0.98与1.22之间。通过polymerchar“仪器控制(instrumentcontrol)”软件以半自动方式制备样品,其中样品重量目标在2mg/ml,且溶剂(含有200ppmbht)经由polymerchar高温自动进样器添加至预先经氮气鼓泡的隔板加盖小瓶中。将癸烷(流动速率标记物)添加至各样品(约5微升)。在“低速”振荡下在160摄氏度下使样品溶解2小时。ir5色谱图基于gpc结果,使用polymerchargpc-ir色谱仪的内部ir5检测器(测量通道)根据等式4-6使用polymerchargpconetm软件(版本2013g)计算mn(conv)、mw(conv)和mz(conv),在各自相等间隔的数据收集点(i)处减去基线的ir色谱图和聚乙烯等效分子量由点(i)的窄标准物校准曲线自等式1获得。表4列举使用等式4-6用于实例和比较实例的常规gpc结果,以下用于常规gpc。为监测随时间的偏差,经由受polymerchargpc-ir系统控制的微型泵将流动速率标记物(癸烷)引入各样品中。流动速率标记物(fm,此处为癸烷)通过使样品内的对应癸烷的rv值(rv(fm样品))对准至窄标准物校准内的癸烷峰的rv值(rv(fm校准)),用以对于各样品线性校正泵流动速率(流动速率(标称))。癸烷标记物峰时间的任何变化随后假定为与整个运行的流动速率(流动速率(有效))中的线性偏移有关。为促进流动标记物峰的rv测量的最高精确性,使用最小平方拟合例程使流动标记物浓度色谱图的峰拟合至二次等式。二次等式的第一导数随后用于求解真实峰位点。在基于流动标记物峰的校准系统之后,有效流动速率(根据窄标准物校准)使用等式7计算。通过polymerchargpconetm软件进行流动标记物峰的处理。可接受流动速率校正为应使得有效流动速率在标称流动速率的+/-2%内。流动速率(有效)=流动速率(标称)*(rv(fm校准)/rv(fm样品))(等式7)。用于测定多检测器偏移量的系统性方法根据由balke,mourey等人公开的方式进行(mourey和balke,《色谱聚合物(chromatographypolym.)》第12章,(1992))(balke,thitiratsakul,lew,cheung,mourey,《色谱聚合物》第13章,(1992))。使用polymerchargpconetm软件进行将三重检测器记录(mw和iv)结果(由宽均聚物聚乙烯标准物(mw/mn=3)产生)对准至窄标准物柱校准结果(由窄标准物校准曲线产生)。光散射色谱图绝对分子量数据(mw(abs))以根据由zimm(zimm,b.h.,《化学物理学期刊(j.chem.phys.)》,16,1099(1948))和kratochvil(kratochvil,p.,《来自聚合物溶液的典型光散射(classicallightscatteringfrompolymersolutions)》,elsevier,牛津(oxford),ny(1987))公开的方式使用polymerchargpconetm软件获得。用于测定分子量的总体注射浓度自质量检测器面积和质量检测器常数获得,其来源于适合的线性聚乙烯均聚物或已知重量平均分子量的聚乙烯标准物中的一种(可追踪至nbs1475均聚物聚乙烯参考样品)。所计算分子量(使用gpconetm)使用来源于如下所述的聚乙烯标准物中的一种或多种的光散射常数,和0.104的折射率浓度系数dn/dc获得。大体而言,质量检测器反应(ir5)和光散射常数(使用gpconetm测定)应由分子量超过约50,000g/mol的线性标准物测定。mw(abs)的等式为使用减去基线的15度光散射信号和减去基线的ir5测量传感器信号(应用质量和光散射常数)的基于面积的结果,如自gpconetm软件所测定,mz(abs)的等式依赖于绝对分子量的逐点测定,其来源于减去基线的15度光散射信号与减去基线的ir5测量传感器信号的比率,且使用gpconetm软件乘以质量常数和光散射常数系数。直线拟合用以外推绝对分子量,其中任一检测器(ir5或ls)在约4%相对峰值信号高度(最大峰值高度)下方。粘度色谱图绝对固有粘度数据(iv(abs))使用自polymerchar粘度计检测器在校准至nbs1475的已知固有粘度时得到的特定粘度色谱图的面积获得。用于测定固有粘度的总体注射浓度自质量检测器面积和质量检测器常数获得,其来源于适合的线性聚乙烯均聚物或已知固有粘度的聚乙烯标准物中的一种(可追踪至nbs1475均聚物聚乙烯参考样品)。iv(abs)的等式为使用减去基线的特定粘度信号(dv)和减去基线的ir5测量传感器信号(应用质量和粘度常数)的基于面积的结果,其由gpconetm软件测定:由三重检测器gpc(tdgpc)的gpcbr分支指数gpcbr分支指数通过首先如先前所描述校准光散射、粘度和浓度检测器来测定。随后自光散射、粘度计和浓度色谱图减去基线。随后设定整合窗以确保使光散射和粘度计色谱图中指示来自折射率色谱图的可检测聚合物的存在的所有低分子量滞留体积范围整合。线性聚乙烯标准物随后用以建立聚乙烯和聚苯乙烯mark-houwink常数。当获得常数时,两个值用以对于聚乙烯分子量和聚乙烯固有粘度作为洗脱体积的函数建构两个线性参考常规校正,如等式(9)和(10)中所展示:gpcbr分支指数为用于表征长链分支的稳健方法,如yau,wallacew.,“3d-gpc-tref用于聚烯烃表征的实例(examplesofusing3d-gpc-trefforpoly-olefincharacterization)”,《大分子聚合物(macromol.symp.)》,2007,257,29-45中所描述。所述指数避免在测定g′值和分支频率计算中传统地所使用的“逐层”tdgpc计算,有利于全部聚合物检测器区域。由tdgpc数据,我们可通过光散射(ls)检测器使用峰面积方法获得样品主体绝对重量平均分子量(mw,abs)。所述方法避免光散射检测器信号相比于浓度检测器信号的“逐层”比率,其如在传统的g′测定中所需要。通过tdgpc,也使用等式(11)独立地获得样品固有粘度。在此情况下面积计算提供更高精确度,因为作为总体样品面积,其对于由检测器噪声和基在线的tdgpc设置和整合限制所产生的偏差的敏感性低很多。更重要地,峰面积计算不受检测器体积偏移量影响。类似地,以等式(11)通过面积方法获得高精确度样品固有粘度(iv):其中dpi表示直接由在线粘度计监测的压差信号。为了测定gpcbr分支指数,样品聚合物的光散射洗脱面积用以测定样品分子量。样品聚合物的粘度检测器洗脱面积用以测定样品的固有粘度(iv或[η])。首先,使用常规校准(“cc”)测定线性聚乙烯标准物样品(如srm1475a或等效物)的分子量和固有粘度,因为分子量和固有粘度均随洗脱体积而变化。等式(13)用以测定gpcbr分支指数:其中[η]为测量的固有粘度,[η]cc为来自常规校准(或convgpc)的固有粘度,mw为测量的重量平均分子量且mw,cc为来自常规校准的重量平均分子量。通过光散射(ls)的重量平均分子量通常称作“绝对重量平均分子量”或“mw(abs)”。使用常规gpc分子量校准曲线(“常规校准”)的mw,cc常常称作“聚合物链主链分子量”、“常规重量平均分子量”和“mw(conv)”。具有“cc或conv”下标的所有统计值使用其对应洗脱体积、如先前所描述的相对应的常规校准和浓度(ci)测定。无下标值为基于质量检测器、lalls和粘度计面积的测量值。kpe的值经反复地调整,直至线性参考样品的gpcbr测量值为0。例如,对于在此特定状况下用于测定gpcbr的α和logk最终值对于聚乙烯分别为0.725和-3.355,且对于聚苯乙烯分别为0.722和-3.993。一旦使用先前论述的程序测定k和α值,则使用支链样品重复程序。使用最终mark-houwink常数作为最佳“cc”校准值来分析支链样品。gpcbr的解释是直接的。对于线性聚合物,gpcbr将接近零,因为通过ls和粘度测定法测量的值将接近常规校正标准。对于支链聚合物,gpcbr将大于0,尤其对于高水平的长链分支,因为测量的聚合物分子量将高于所计算的mw,cc且所计算的ivcc将高于测量的聚合物iv。实际上,gpcbr值表示因作为聚合物分支的结果的分子尺寸收缩作用所致的iv变化分数。gpcbr值为0.5或2.0意指iv的分子尺寸收缩作用分别处在相对于线性聚合物分子当量50%和200%的水平。对于这些特定实例,与传统的“g′指数”和分支频率计算相比使用gpcbr的优势是由于gpcbr的较高精确度。在gpcbr指数测定中使用的所有参数以良好精确度获得,且未不利地受处于来自浓度检测器的高分子量的低tdgpc检测器反应影响。检测器体积对准中的错误也不会影响gpcbr指数测定的精确度。计算lcb频率(lcbf)通过以下程序对于各聚合物样品计算lcbf:1)用nbs1475均聚物聚乙烯(或等效参考)校准光散射、粘度和浓度检测器。2)相对于如上文在校准部分中所述的浓度检测器校正光散射和粘度计检测器偏移量(参见对mourey和balke的引用)。3)自光散射、粘度计和浓度色谱图减去基线且设定整合窗以确保使可自折射计色谱图观测的在光散射色谱图中的所有低分子量滞留体积范围整合。4)通过注入多分散性为至少3.0的标准物确认线性均聚物聚乙烯mark-houwink参考线,计算数据文件(来自上文的校准方法)且对于各色谱图块记录来自质量恒定经校正数据的固有粘度和分子量。5)分析所关注的ldpe样品,计算数据文件(来自上文的校准方法)且对于各色谱图块记录来自质量恒定经校正数据的固有粘度和分子量。在较低分子量下,可能需要将固有粘度和分子量数据外推使得测量的分子量和固有粘度渐近地接近线性均聚物gpc校准曲线。6)均聚物线性参考固有粘度在各点(i)处通过以下因子转换:ivi=ivi*0.964,其中iv为固有粘度。7)均聚物线性参考分子量通过以下因子转换:m=m*1.037,其中m为分子量。8)各色谱图块处的g′根据以下等式计算:g′=(iv(ldpe)/iv(线性参考)),在相同m下。自参考mark-houwink曲线拟合的五阶多项式计算iv(线性参考)且其中iv(线性参考)为线性均聚物聚乙烯参考物的固有粘度(添加一定量scb(短链分支)以造成在相同分子量(m)下通过6)和7)尾咬)。iv比率假定为在小于3,500g/mol的分子量下以造成光散射数据中的天然散射。9)根据以下等式计算在各数据图块处的分支数目:10)平均lcb数量根据以下等式(lcb1000c=lcbf)在所有图块(i)中计算:分子架构测定为测定各种聚合物组合物的分子架构,使用以下程序。色谱系统由配备有4个毛细管粘度计和agilenttechnologies双角度雷射光散射检测器型号2040的polymerchargpc-ir高温色谱组成。光散射检测器的15度角度用于计算目的,且ir5“测量通道”用以测量浓度。收集且使用polymerchargpc软件处理数据。系统配备有在线溶剂脱气装置。在160℃下操作柱隔室。所用柱为4agilent“mixeda”30cm20微米分析柱。所用溶剂为1,2,4-三氯苯。以50毫升的溶剂中0.1克的聚合物的浓度制备样品。色谱溶剂和样品制备溶剂含有200ppm的丁基化羟基甲苯(bht)。两种溶剂源均经氮气鼓泡。将聚乙烯样品在160摄氏度下轻轻摇晃(速度为1)3小时。所用注射体积为200微升且流动速率为1.0ml/min。通过分子量在580至8,400,000g/mol范围内的至少20种窄分子量分布聚苯乙烯标准品进行gpc柱组的校准,且所述标准品以6种“混合液”混合物形式组态,个别分子量之间相差大约十倍。标准品购自agilenttechnologies。对于分子量等于或大于1,000,000,以在50毫升溶剂中0.025克、且对于分子量小于1,000,000,以在50毫升溶剂中0.05克来制备聚苯乙烯标准品。通过在80℃下轻轻搅拌30分钟预溶解聚苯乙烯标准品。将聚苯乙烯标准品峰分子量使用以下等式(如williams和ward,《聚合物科学期刊(j.polym.sci.)》,《聚合物快报》,6,621(1968))中所描述)转换为聚乙烯分子量:m聚乙烯=a×(m聚苯乙烯)b,其中m为分子量,a的值为0.41且b等于1.0。三阶多项式用于拟合对应聚乙烯当量gpclog(分子量)校准点。通过癸烷(以在50毫升tcb中0.04g制备,且通过轻轻搅拌溶解20分钟)进行gpc柱组的总板计数。板计数和对称性根据以下等式在200微升注射上进行测量。其中rv为以毫升为单位的滞留体积。其中rv以毫升为单位。色谱系统的板计数必须高于22,000且对称性必须低于1.25。测定多检测器偏移量的系统方法以与balke,mourey,等人(mourey和balke,《色谱聚合物》第12章,(1992))(balke,thitiratsakul,lew,cheung,mourey,《色谱聚合物》第13章,(1992))出版的一致的方式进行,使用内部软件优化来自陶氏广泛聚苯乙烯1683的双重检测器记录结果至来自窄标准品校准曲线的窄标准品柱校准结果。偏移测量定的分子量数据以与zimm(zimm,b.h.,j.chem.phys.,16,1099(1948))和kratochvil(kratochvil,p.,《来自聚合物溶液的经典光散射(classicallightscatteringfrompolymersolutions)》,elsevier,oxford,ny(1987))出版的一致的方式获得。用于测定分子量的总注射浓度自样品折射率面积和来自120,000分子量的直链聚乙烯均聚物的红外检测器校准中获得。假定色谱浓度低至足以消除针对第2维里系数(virialcoefficient)的效应(针对分子量的浓度效应)。通过直链均聚物参考材料的光散射所得的log(分子量)洗脱应与如上文所描述的常规gpc一致。癸烷包含(通过gpc-ir微型泵)于各校准和样品流动中,且用于提供参考各样品流回至初始校准曲线的流动速率。用于红外测量通道(“cdfir”)粘度计检测器(“cdfdv”)和低角度雷射光散射检测器(“cdfls”)的累积检测器份数(cdf)的计算通过以下步骤实现:1)基于样品与一致窄标准品混合液混合物之间的空气峰的相对滞留体积比,线性流修正色谱图。2)相对于如校准部分中所描述的折射计,修正光散射检测器和粘度计检测器偏移量。3)自光散射、粘度计和折射计色谱图减去基线且设定整合窗以确保使自折射计色谱图可观测的光散射和粘度计色谱图内的所有低分子量滞留体积范围整合。4)基于经如校准部分中所描述的聚苯乙烯至聚乙烯转换因子(0.41)修改的聚苯乙烯校准曲线计算各数据层下的分子量。5)以如以下其滞留体积所描述的两个所需gpc分子量点之间的色谱面积计算各色谱图(adfir、adfdv和adfls)的面积检测器份数(adf):因此,adf(adfir、adfdv和adfls)定义为在gpc分子量所需范围内的“反应(强度)乘滞留体积”中经整合的色谱图的面积除以经整合的色谱图的全部面积。如果所需分子量在色谱图的经整合的面积之外,则所述点以外的所需分子量的任何面积层相当于零;因此adf分子表示所需范围与完全色谱经整合的面积范围的相交。同样,累积检测器份数(cdf)相较于分子量的曲线图可通过在各经整合的滞留体积(i)下计算自最高分子量限制(最低经整合的滞留体积)至各经整合的滞留体积直至达至最低分子量限制的adf而获得。以此方式,cdf可自0至1绘制,且所需色谱图的面积份数(adf)可以两个cdf值之间的差读取。因此,cdfi为gpc分子量≥(或≤)表现为gpc分子量的所需值的全部经整合的色谱面积的份数。所需组合物在≤15,000g/molgpc分子量下的adfir(或adfir)为0.200至0.250。图1展示具有0.222的值的本发明实例1。此意味着所有本发明组合物在分子量≤15,000g/molgpc分子量下展示20%的浓度分布,其使树脂在所关注的mi范围内具有极佳可处理性。adfir≤0.250(为本发明聚合物而展示)限制“可萃取物”可产生问题的低分子量材料的量。高分子量材料必须进一步具有足够长分支链臂,其通过光散射检测器良好地检测。因此我们需要足够大的极高分子量份数(在至少0.350的≥500,000g/molgpc分子量下adfls(或adfls))。因此35%的光散射色谱图在分子量≥500,000g/molgpc分子量下尤其有利。值得注意的,极高分支材料的量有限制,且据相信,对于分子量≥500,000g/molgpc分子量,此材料应保持≤45%(adfls≤0.450)在此特定mi(i2)范围(1.5-3.0g/10min)中以避免凝胶的并发作用,其可影响透明度、表面纹理和可拉性。在≥500,000g/molgpc分子量下,最佳adfls为0.350至0.450(图2)。图2中呈现的本发明实例1产生0.376的adfls值。此外,指示全部主链和分支分子量的通过光散射的绝对分子量(mw(abs))目标范围处于130,000至162,000g/mol。此值可直接由经校准的15度光散射通道的浓度标准化面积获得。固有粘度(iv)指示更大、更多直链的量,且优选应保持相对较低,低于0.880dl/g,或更优选在0.820至0.880dl/g范围内(如通过线内gpc粘度计所测量)以用于最佳处理。因为有必要维持某些长链以增强树脂熔融强度,且粘度计可用于判定是否存在足够的该等材料,所以我们检查在≥200,000g/molgpc分子量下的dv的累积检测器份数。此表明链充分足够长以用于缠结,但一般在可造成光学上的困难程度上不交联。我们已发现在分子量≥200,000g/mol下具有至少0.180的adfdv(或adfdv)份数的聚合物分子在所关注的mi(i2熔融指数)范围内(图3)足够用于此准则(至少18%的粘度计色谱图表示分子量≥200,000g/mol(gpc分子量))。对于本发明实例1,此测定展示为adfdv的0.197。差示扫描热测量定(dsc)可使用差示扫描量热法(dsc)以测量聚合物在广泛范围温度内的熔融和结晶行为。举例而言,将装备有rcs(冷冻冷却系统)和自动进样器的tainstrumentsq2000dsc用于执行此分析。在测试期间,使用50ml/min的氮气冲洗气体流。将各样品在约190℃下熔融压成薄膜;接着使熔融样品空气冷却至室温(约25℃)。通过“0.5至0.9克”样品在190℃下以20,000lbf按压且按压10秒以形成“0.1至0.2密耳厚度”膜,来形成膜样品。3-10mg、6mm直径样本自冷却聚合物萃取,称重,置放于轻质铝盘(约50mg)中,且卷曲关闭。随后进行分析,以测定其热性质。样品的热行为通过缓慢升高且降低样品温度以建立相对于温度分布的热流动来测定。首先,将样品快速加热至180℃,且保持等温5分钟,以便去除其热历程。随后,使样品以10℃/分钟冷却速率冷却至-40℃,且保持等温在-40℃下5分钟。随后将样品以10℃/分钟加热速率加热至150℃(此为“第二加热”匀变)。记录冷却和第二加热曲线。通过自结晶的起始设定基线端点至-20℃分析冷却曲线。通过自-20℃设定基线端点至熔融终点分析加热曲线。测定的值为峰值熔融温度(tm)、峰值结晶温度(tc)、熔化热(hf)(以焦耳每克为单位),且使用以下等式计算乙烯类聚合物样品的%结晶度:%结晶度=((hf)/(292j/g))×100(等式14)。熔化热(hf)和峰熔融温度由第二加热曲线报告。峰值结晶温度由冷却曲线测定。膜测试以下物理特性在实验部分中所描述的膜上测量。在测试之前,使膜在23℃(+/-2℃)下和50%相对湿度(+/-10%相对湿度(r.h))下适应至少40小时(在膜制造之后)。各膜的厚度参见表12。总(总体)混浊度和内部混浊度:根据astmd1003测量内部混浊度和总混浊度。使用矿物油(1-2茶匙)经由折射率匹配内部混浊度,将所述矿物油涂覆为在各膜表面上的涂层。ahazeguardplus(马里兰州哥伦比亚的美国毕克-加特纳(byk-gardnerusa;columbia,md))用于测试。对于各测试,检测5个样品且报告平均值。样品“6英寸×6英寸”。45°光泽:astmd2457(五个膜样品的平均值;各样品“10英寸×10英寸”)。透明度:使用上文的ahazegardplus设备测量(五个膜样品的平均值;各样品“10英寸×10英寸”)。zebedee透明度:astmd1746使用zebedee透明度计型号cl-100(五个膜样品的平均值;各样品“4.5英寸×4.5英寸”)。2%正割模数-md(纵向)和cd(横向):astmd882(在各方向上5个膜样品的平均值)。将1英寸宽的测试条使用线接触夹具在间隔4英寸的接触点(标距)处装载在抗张测试支架中。样品以2英寸/分钟的十字头速度至多标称染色的5%测试。md和cd艾勉道夫(elmendorf)撕裂强度:astmd1922。在整个膜或薄片样本中传播撕裂所需要的力(以克为单位)使用精确校准的摆锤装置来测量。受重力影响,摆锤沿弧线摆动,自预先切割的缝隙撕裂样品。试样在一侧上通过摆锤固定且在另一侧上通过固定构件固定。通过指示器或通过电子秤指示摆锤所导致的能量损失。所述秤示数为撕裂样品所需的力的函数。所使用的样品为如d1922中所指定的‘恒定半径几何体’。测试通常将在已沿md和cd方向切割的样品上进行。测试之前,在样品中央处测量膜样品厚度。每方向测试总共15个样本,且报告平均撕裂强度。在自垂直大于60°的角度下撕裂的样品描述为‘倾斜’撕裂-应标注此类撕裂,但强度值包含于平均强度计算中。穿孔强度:在instron型号4201上用sintechtestworks软件版本3.10测量穿孔。样本尺寸为“6英寸×6英寸”且进行5次测量以测定平均穿孔值。使用具有4英寸直径的圆形样本固持器的“100lb测力计”。穿孔探针为在0.25英寸直径支撑棒上的1/2英寸直径、经抛光不锈钢滚珠,其最大行进长度为7.5英寸。不存在标距,且在测试开始之前探针尽可能地接近但不接触样本。将穿孔探针以10英寸/分钟的十字头速度推至经夹持的膜的中央。在试样的中央处进行单一厚度测量。对于各样本,测定穿孔((ft·lbf)每英寸3)。在各样本之后使用“金佰利拭纸(kim-wipe)”对穿孔探针进行清洁。“低收缩力膜的收缩力测量”,speantec论文集,第1264页(2008)。通过在具有膜夹具的rsa-iii动力学机械分析器(tainstruments;newcastle,de)上进行的温度斜坡测试来测量膜样品的收缩张力。自膜样品以纵向(md)或横向(cd)模切“12.7mm宽”和“63.5mm长”的膜样本,用于测试。膜厚度通过mitutoyo绝对电子数显(digimatic)指示器(型号c112cexb)测量。此指示器的最大测量范围为12.7mm,分辨率为0.001mm。在各膜样本上的不同位置处的3次厚度测量的平均值和样本的宽度用以计算膜的横截面积(a),其中在收缩膜测试中所使用的膜样本的“a=宽度×厚度”。使用tainstruments的标准膜张力夹具。在将间隙和轴向力调零之前rsa-iii的烘箱在25℃下平衡至少30分钟。初始间隙设定为20mm。随后将膜样本连接在夹具的上部和下部。通常,对于md的测量仅需要一层膜。因为cd方向上的收缩张力通常为低的,将两层或四层膜堆栈在一起用于各测量,以改良信号对噪声比。在此情况下,膜厚度为所有层的总和。在此工作中,在md方向上使用单一层且在cd方向上使用两层。在膜达到25℃的初始温度之后,人工地使夹具上部略微升高或降低以获得-1.0g的轴向力。此确保在测试开始处不发生膜的屈曲或过度拉伸。随后开始测试。在整个测量期间保持恒定夹具间隙。温度斜坡以90℃/min的速率开始自25℃至80℃,随后以20℃/min的速率自80℃至160℃。在来80℃至160℃的斜坡期间,在膜收缩时将通过力转换器测量的收缩力记录为温度的函数用于分析。“峰值力”与“在收缩力峰值开始之前的基线值”之间的差值视为膜的收缩力(f)。膜的收缩张力为收缩力(f)比膜的初始横截面积(a)的比率。实验制备本发明乙烯类聚合物图4为用以产生本发明乙烯类聚合物(ldpe)的工艺反应系统的方块图。图4中的工艺反应系统为部分闭合回路、双再循环、高压、低密度聚乙烯生产系统。工艺反应系统由以下组成:新鲜乙烯进料线[1]、增压器和主压缩机(“主”)、超压缩机(“超”)和四区管状反应器(“4区反应器”)。通过“预加热器”将物流[3]加热至足够高的温度且馈入至反应器前部。将物流[11]作为侧流馈入至反应器中。在反应器中,借助于四种混合物引发聚合,各含有一种或多种自由基引发系统(参见表1),其在各反应区(图中未示)的入口处注入。通过调节各反应区开始处引发剂的混合物的馈入量将各反应区中的最大温度控制于设定点。各反应区具有一个入口和一个出口。各入口流由来自前述区的出口流和/或所添加的富乙烯进料流组成。当聚合完成时,反应混合物以物流[4]减压且冷却。工艺进一步由高压分离器“hps”组成,其将反应混合物分离成富乙烯流[8](所述富乙烯流[8]经冷却和再循环回到超的吸入口)和富聚合物流[5](其递送至低压分离器“lps”用于进一步分离)。在lps中,使富乙烯流冷却且以物流[6]再循环回到增压器(“增压器”)。乙烯自增压器进一步通过主压缩机压缩。进料[2]随后再循环至超压缩机的吸入口。离开lps[7]的聚合物进一步进行粒化和冲洗。将链转移剂“cta”馈料[10]注入至主压缩机排出口的乙烯流中。物流[9]为用以去除杂质和/或惰性物质的净化流。将冷却夹套(使用高压水)安装在管反应器和预加热器的外壳周围。对于本发明实例1-2,含有过氧-2乙基己酸叔丁酯(tbpo)、过氧乙酸叔丁酯(tbpa)和异石蜡烃溶剂的混合物(沸腾范围171-191℃;例如isoparh)用作第一反应区的引发剂混合物。对于第二反应区,使用含有二叔丁基过氧化物(dtbp)、tbpo、tbpa和异石蜡烃溶剂的混合物。对于第三和第四反应区,使用tbpa、dtbp和异石蜡烃溶剂的混合物。此数据概括于表1中。1-丁烯用作cta。馈入至工艺的cta的浓度经调节以控制产品的熔融指数。表2显示用以形成实例1和2的聚合条件。表1:对于实例1和2在各注入点处的过氧化物引发剂流,以磅每小时为单位注入点引发剂实例1:纯po磅/小时实例2:纯po磅/小时#1tbpo1.871.80#1tbpa0.800.77#2tbpo5.45.20#2tbpa1.161.11#2dtbp1.161.11#3tbpa00#3dtbp00#4tbpa3.043.10#4dtbp7.107.24表2:工艺条件实例1实例2反应器压强(psig)38,71838,8981区起始t(℃)146.5148.71区峰值(℃)251.6247.02区起始t(℃)163.6168.62区峰值t(℃)288.7286.63区起始t(℃)246.4246.23区峰值t(℃)240.2239.24区起始t(℃)196.3191.94区峰值t(℃)283.2281.0新鲜乙烯流(lb/hr)23,84025,332至反应器的乙烯吞吐量(lb/hr)100,480100,600乙烯转化率(%)23.724.2丁烯流(lb/hr)223245乙烯冲洗流(lb/hr)509505再循环丁烯浓度(体积%)0.540.59预加热器t(℃)200.3200.3反应器冷却系统1(℃)168.4167.9反应器冷却系统2(℃)176.8176.5本发明实例和比较实例的性质列出于表格3-10中。表3含有熔融指数(i2或mi)、密度、%己烷可萃取物、熔融强度和熔融强度数据的断裂时速度。在样品中任一个的最高密度为0.925g/cc的情况下本发明实例在熔融指数2至3的范围内,表明这些ldpe树脂将具有改良的硬度。此外,本发明实例的己烷可萃取物极低,处于0.9-1.0重量%,表明在使用时可来ldpe萃取较少材料。如图5中所示本发明实例的熔融强度相对较低,且在190℃下熔融强度为<-1.87*(190℃下i2)+10cn。此外,本发明实例的断裂时速度为高的,表明良好可拉性和挤压行为。表格4至表6含有本发明实例和比较聚合物的tdgpc数据。表4含有常规gpc(conv)数据其说明由所适当宽mw(conv)/mn(conv)比率描述的适当宽分子量分布,与适当高的z-平均分子量、mz(conv)结合,同时具有较低重量平均分子量、mw(conv),以上所有有助于熔融强度和吹制膜生产在线产量的良好平衡,如对于这些本发明聚合物所见。表5含有来源于ls和粘度检测器以及浓度检测器的tdgpc相关的性质。如表5中所见,已发现本发明聚合物具有较高mz(abs)/mw(abs)比率,伴随着相对较低mw(abs)。此外,lcbf和gpcbr值突出本发明树脂具有相对较低量的长链分支,且因此具有更“开放结构”,允许高分子量mz(abs)分子更有机会互相渗透且有效地改进熔融强度。表6含有三重检测器色谱图的特征,包含有助于加工的低分子量尾部(由adfir定义)、有效量的高分子量支链物种(其特征在于适中adfls值)和特定粘度色谱图(adfdv)的极低部分(其将由高mw线性物种(较高粘度和较低熔融强度)控制)。此通过自其计算固有粘度(iv(abs))的特定粘度色谱图的总面积突出,对于这些聚合物所述固有粘度典型地为低的。其为低粘度、iv(abs)(尤其在高mw区域中)、adfdv的平衡,其允许极好的加工同时仍维持前述特征的所期望的水平(表5和adfls),产生良好熔融强度且为本发明实例的特征。表7含有如通过以下概述的dms粘度数据:在0.1、1、10和100rad/s下所测量的粘度,粘度比或在0.1rad/s下测量的粘度比在100rad/s下测量的粘度的比率(其皆在190℃下测量)和在0.1rad/s和190℃下测量的正切δ。对于本发明聚合物,反映粘度随着频率的变化的粘度比相对较低。本发明聚合物在0.1rad/s下的正切δ值相对较高,指示低熔融弹性或熔融强度。表8含有如通过13cnmr所测量的每1000总碳原子的分支。这些ldpe聚合物含有大致上不包括于线性聚乙烯或lldpe中的戊基或c5分支,所述线性聚乙烯如affinity聚烯烃塑性体,所述lldpe如dowlex聚乙烯树脂,其均由陶氏化学公司制造。展示于表8中的各本发明和比较ldpe含有大于或等于每1000总碳原子0.5个戊基基团(分支)(本发明实例含有大于每1000总碳原子1个戊基基团(分支))。本发明实例含有每1000总碳原子相对不可检测的c1。表9含有通过1hnmr的不饱和结果。表10含有熔点、tm、熔化热、%结晶度和结晶点tc的dsc结果。表3:熔融指数(i2)、密度、%己烷可萃取物、190℃下的熔融强度(ms)和实例(ex.)和比较实例(ce)的熔融强度的断裂时速度(v)*可自陶氏化学公司获得。**可自braskem获得。表4:实例(ex.)和比较实例(ce)的常规gpc性质mn(conv)(g/mol)mw(conv)(g/mol)mz(conv)(g/mol)mw(conv)/mn(conv)ex.115,60081,100283,0005.18ex.215,40079,300282,0005.16ce114,10099,400376,0007.06ce217,10089,400299,0005.21ce315,50093,500323,0006.03ce414,70090,900382,0006.20ce512,900101,000448,0007.81ce615,70078,900318,0005.03ce717,60078,800271,0004.48ce817,70080,900224,0004.57表5:实例(ex.)和比较实例(ce)的绝对gpc校准。表6:实例(ex.)和ce的tdgpc性质(ir、ls和dv)表7:在0.1、1、10和100rad/s下以pa·s为单位的粘度,粘度比,和190℃下的正切δ表8:实例和比较实例通过13cnmr的每1000c分支*未检测到。表9:实例和比较实例通过1hnmr的不饱和结果表10:实例和比较实例的dsc结果样品tm(℃)熔化热(j/g)%结晶度tc(℃)ex.1112.0154.552.9100.8ex.2112.0153.052.4100.8ce1112.0150.451.5100.9ce2110.3146.150.098.8ce3110.0134.946.296.8ce4108.5140.348.096.4ce5108.3136.246.695.9ce6108.2145.349.895.8ce7115.4169.257.9103.1ce8109.1149.551.297.7用不同ldpe制成吹制膜且测量物理特性。膜以100wt%ldpe制成。单层吹制膜在“8英寸模”上用聚乙烯“davis标准屏障型ii螺杆”制成。通过空气环外部冷却且使用内部鼓泡冷却。用以制造各吹制膜的通用吹制膜参数展示于表11中。温度为最接近丸粒料斗(筒1)的温度,且在将聚合物挤压通过模时为增加的顺序。表11:用于膜的吹制膜制造条件。膜性质展示于表12中。本发明样品具有如所期望的良好混浊度、光泽和透明度和高2%md正割模数,反映良好硬度。本发明组合物提供adfir值、分子量分布和熔融指数(i2)的适当平衡,且良好加工成吹制膜和收缩膜。如果乙烯类聚合物的adfls值<0.350(参见膜10),则对于所要求的i2范围我们发现cd收缩张力降低和穿孔耐性降低。如果乙烯类聚合物的adfls值>0.450,则聚合物将具有过多的高分子量含量,其通常引起凝胶水准增加和较差光学(光泽降低)。如果mw(abs)<130,000g/mol,则通常产生降低的熔融强度,引起鼓泡强度降低。如果mw(abs)>162,000g/mol,产生过多高分子量含量,其可导致模量降低和cd收缩张力(或硬度-参见膜4、5和8)降低和/或膜产品中非所期望的结晶或定向。如果聚合物的i2值<1.5,则通常较难以将聚合物挤压成吹制膜,且通常在膜产品中引起不良光学(混浊度增加且光泽降低)。如果聚合物的i2值>3.0,此将通常使得最终膜收缩张力较低,且由此引起膜中的松弛增加,且丧失围绕所容纳物品的包装紧密性。参见膜9-cd收缩张力的降低,穿孔耐性的降低和混浊度的增加(不良光学)。表12:各自以2.0密耳(±0.3)在约250lb/hr的标准(std.)速率下制成的“100%ldpe”膜#1-10的膜性质当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1