一种吡啶三唑修饰的香豆素Cu2+荧光探针的制备方法与流程
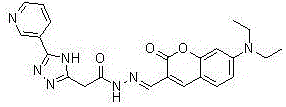
本发明涉及一种吡啶三唑修饰的香豆素cu2+荧光探针的制备方法,具体属于有机合成技术领域。
背景技术:
金属离子对环境和人体健康具有重要的影响,因此检测环境中金属离子的方法受到了各研究人员的广泛关注。检测金属离子的方法很多,如m.-s.chan,s.-d.huang在“directdeterminationofcadmiumandcopperinseawaterusingatransverselyheatedgraphitefurnaceatomicabsorptionspectrometerwithzeeman-effectbackgroundcorrector”(talanta51(2000)373–380.)中,描述了原子吸收光谱法;p.fodor,b.d.zs在“uncertaintyinenvironmentalicp-aesmeasurements”(microchem.j.51(1995)151–158.)中阐述的等离子体原子发射光谱法;j.wu,e.a.boyle在“lowblankpreconcentrationtechniqueforthedeterminationoflead,copper,andcadmiuminsmall-volumeseawatersamplesbyisotopedilutionicpms”(anal.chem.69(1997)2464–2470.)中涉及了质谱法;j.homola,s.s.yee等人,在“surfaceplasmonresonancesensors”(review,sens.actuatorsb54(1999)3–15.)提到表面平面共振等。与上述传统方法相比较,基于金属离子检测的荧光探针具有高灵敏性、专一性、可实时检测性和成本低等优点,因而成为目前研究的热点。
荧光探针是以荧光物质作为指示剂,并在一定波长光的激发下使指示剂产生荧光,通过检测所产生的荧光,实现对被检测物质的定量或定性分析。荧光分子探针通常由识别基团、荧光基团、连接体三部分组成。识别基团决定了探针分子的选择性和特异性,荧光基团则决定了识别的灵敏度,而连接体部分则可起到分子识别枢纽的作用。目前已有报道基于cu(ii)选择性检测的荧光化学传感器,并已在生物应用中取得了一定应用,然而,这些手段中也存在实际应用的不足,例如配体的细胞毒性,小的斯托克斯位移,响应缓慢[6]对其他金属离子的交叉敏感性,水性介质中的低荧光量子产率。因此,有必要找到一种可以克服这些缺点的荧光探针。
目前,制备检测铜离子的荧光探针的现有技术路线主要有两种,具体如下所述:
技术路线1
l.zeng,e.w.miller等人,在“aselectiveturn-onfluorescentsensorforimagingcopperinlivingcells”(j.am.chem.soc.128(2006)10–11.)中提到技术路线1。如技术路线1所示:bodipy(1)通过2,4-二甲基-3-乙基吡咯与氯乙酰氯缩合,然后用bf3·oet2处理,以一锅两步法获得,两步的总收率为16%;四硫代受体(3)分两步递送,乙基2-羟乙基硫醚与硫脲和hbr的转化顺利进行,产生硫醇(2),产率84%;在碱性条件下用双(2-氯乙基)胺盐酸盐处理硫醇(2)得到产物四硫代受体(3),产率为79%;在回流的乙腈中偶联bodipy(1)和四硫代受体(3),再经后处理和纯化后得到cs1(4),产率为22%。该路线的缺点是合成路线繁琐,收率偏低。
技术路线2:
b.gu,l.y.huang,w.su等人,在“abenzothiazole-basedfluorescentprobefordistinguishingandbioimagingofhg2+andcu2+”(anal.chim.acta954(2017)97–104.)中描述了技术路线2。如技术路线2所示:以化合物(1)(3.00g,19.98mmol)和1,3-丙二硫醇(2.38g,21.98mmol)为原料,以三氟化硼二乙基醚合物为催化剂,二氯甲烷为溶剂,制得化合物(2);然后以化合物(2)与氨基苯硫酚为原料,氨基磺酸为催化剂,水为溶剂,制备化合物(bt)。该路线的缺点是响应慢,需要8min。
技术实现要素:
针对前面技术中存在的诸多不足,本发明的主要目的在于提供一种能快速检测铜离子的荧光探针及其相关制备方法,合成了一种基于吡啶三唑修饰的香豆素cu2+荧光探针。
本发明3-甲酰基-7-(二乙氨基)香豆素席夫碱的制备方程式分别为:
本发明一种吡啶三唑修饰的香豆素cu2+荧光探针的制备方法,所述的吡啶三唑修饰的香豆素cu2+荧光探针为3-甲酰基-7-(二乙氨基)香豆素席夫碱,其结构式为:
其制备方法包括以下步骤:
步骤1:化合物a的制备
控制4-(二乙基氨基)水杨醛、丙二酸二乙酯、哌啶的当量比为1∶2∶1,将4-(二乙基氨基)水杨醛溶于乙醇中,加入丙二酸二乙酯和哌啶,加热搅拌至回流12小时;通过tlc监测反应完毕后,减压除去产物中的溶剂,然后加入体积比为1∶1的浓盐酸和冰醋酸进行水解,搅拌反应6小时;水解产物冷却后倒入冰水中,再加入40wt%naoh水溶液调节溶液ph至7,向其中加入二氯甲烷进行萃取,有机相再经无水mgso4干燥后旋蒸浓缩,通过柱色谱用乙酸乙酯/正己烷(体积比为1∶3)纯化,得到4-(二乙氨基)香豆素;
控制dmf、pocl3、4-(二乙氨基)香豆素的当量比为3∶2.5∶1,在50℃、ar保护下,将无水dmf滴加到pocl3中并搅拌30分钟,得到红色溶液;将4-(二乙氨基)香豆素的dmf溶液通过恒压滴液漏斗缓慢滴加到上述红色溶液中,然后在70℃条件下搅拌反应12小时,通过tlc监测反应完毕后,恢复室温条件,将反应液倒入冰水中,加入20wt%naoh水溶液调节溶液ph至7,其后用乙酸乙酯分批萃取,合并有机相后用饱和食盐水洗涤3次,有机相再经无水mgso4干燥后旋蒸浓缩,通过柱色谱用乙酸乙酯/正己烷(体积比为1∶2)纯化,得到化合物(a);
步骤2:化合物(b)的制备
控制化合物(i)、hcl(g)的当量比为1∶1.2,将干燥的氯化氢用鼓泡法溶于无水乙醇溶剂中,滴加2-氰基乙酸乙酯,在0℃下反应4小时后有白色沉淀析出,减压旋蒸除去溶剂乙醇,析出的白色晶体用石油醚重结晶,得到化合物(ii);
控制化合物(iii)、无水乙醇、浓硫酸的当量比为40∶250∶1,将化合物(iii)溶于无水乙醇中,滴加浓硫酸,搅拌反应混合物并加热至回流4小时,通过tlc监测反应完毕后,减压除去溶剂,再通过柱色谱用乙酸乙酯/正己烷(体积比为1∶3)纯化得到化合物(iv);
控制化合物(vi)、水合肼的当量比为1∶2,将化合物(iv)溶于乙醇中,加入水合肼,搅拌反应混合物并加热至回流过夜;通过tlc监测反应完毕后,减压除去溶剂,析出的白色晶体用乙醇重结晶,得到化合物(v);
控制化合物(ii)、化合物(v)的当量比为2∶1,将化合物(ii)溶于乙醇中,加入化合物(v),搅拌反应混合物并加热至回流36小时,然后冷却至室温;用乙酸乙酯进行萃取,有机层分别用水洗涤3次;通过柱色谱用的甲醇/乙酸乙酯(体积比为1∶30)纯化,并真空干燥,得到化合物(vi);
控制化合物(vi)、水合肼的当量比为1∶2,将化合物(vi)溶于乙醇中,加入水合肼,搅拌反应混合物并加热至回流过夜;通过tlc监测反应完毕后,减压除去溶剂乙醇,析出的白色晶体用乙醇重结晶,得到化合物(b);
步骤3:化合物(c)3-甲酰基-7-(二乙氨基)香豆素席夫碱的制备
控制化合物(a)、化合物(b)的当量比为1∶1,将化合物(a)溶于乙醇中,加入化合物(b),搅拌反应混合物并加热至回流过夜,通过tlc监测反应完毕后,减压除去溶剂乙醇,析出的橙红色晶体用乙醇重结晶,得到化合物(c)3-甲酰基-7-(二乙氨基)香豆素席夫碱。
本发明的有益效果:本发明3-甲酰基-7-(二乙氨基)香豆素作为检测cu2+的荧光探针,具有光稳定性好、荧光量子产率高、斯托克位移大等优点,能快速、高效、专一的识别水溶液中的铜离子,是一种快速高灵敏性的铜离子荧光探针,且对其他常见金属离子具有较强的抗干扰能力,实时监测及检测稳定性高,可用来定性、定量检测环境中的铜离子,对铜离子的检测具有很好的应用价值。本发明反应条件易控,操作方便,产物收率高。本专利申请系国家自然科学基金项目(21501088)支持。
附图说明
图1为本发明3-甲酰基-7-(二乙氨基)香豆素席夫碱的结构式;
图2为本发明3-甲酰基-7-(二乙氨基)香豆素席夫碱的荧光光谱图;
图3为本发明3-甲酰基-7-(二乙氨基)香豆素席夫碱的荧光滴定实验图;
图4为本发明3-甲酰基-7-(二乙氨基)香豆素席夫碱的离子竞争实验图;
图5为本发明3-甲酰基-7-(二乙氨基)香豆素席夫碱的核磁共振氢谱图。
具体实施方式
下面结合实施例对本发明作进一步详细的描述,以令本领域的技术人员参照说明书文字能够据以实施。
实施例1
检测铜离子的荧光探针为3-甲酰基-7-(二乙氨基)香豆素席夫碱,制备过程具体为:
步骤1:化合物a的制备
将4-(二乙基氨基)水杨醛1.93g溶于40.0ml乙醇中,加入丙二酸二乙酯3.12ml和哌啶1ml,加热搅拌至回流过夜;通过tlc监测反应完毕后,减压除去产物中的溶剂,然后加入浓盐酸20.0ml和冰醋酸20.0ml进行水解并再搅拌6小时;水解产物冷却后倒入100ml冰水中,再加入40wt%naoh水溶液调节溶液的ph至7;向其中加入30.0ml二氯甲烷进行萃取,有机相再经无水mgso4干燥后旋蒸浓缩,通过柱色谱用乙酸乙酯/正己烷(体积比为1∶3)纯化,得到2.01g4-(二乙氨基)香豆素,收率93%;
在50℃、ar保护下,将干燥的dmf2.14ml滴加到pocl32.18ml中并搅拌30分钟,得到红色溶液;将2.01g4-(二乙氨基)香豆素溶于10.0mldmf中,并通过恒压滴液漏斗缓慢滴加到红色溶液中,在70℃下搅拌反应12小时,通过tlc监测反应完毕后,恢复室温条件,然后将反应液倒入100ml冰水中,加入20wt%naoh水溶液调节溶液的ph至7;其后用30.0ml乙酸乙酯分批萃取,合并有机相后用饱和食盐水洗涤3次,有机相再经用无水mgso4干燥后旋蒸浓缩,通过柱色谱用乙酸乙酯/正己烷(体积比为1:2)纯化,得到化合物(a)1.72g,收率76%。
步骤2:化合物(b)的制备
将干燥的氯化氢用鼓泡法溶于无水乙醇溶剂中,滴加5.0g2-氰基乙酸乙酯。在0℃下反应4小时后有白色沉淀析出。减压旋蒸除去溶剂,析出的白色晶体用石油醚重结晶,得到化合物(ii)5.83g。收率83%。
将化合物(iii)6.0g溶于50.0ml无水乙醇中,滴加2滴浓硫酸,搅拌反应混合物并加热至回流4小时,通过tlc监测反应完毕后,减压除去溶剂,再经用柱色谱用乙酸乙酯/正己烷(1:3)纯化,得到化合物(iv)5.96g,收率81%。
将化合物(iv)5.96g溶于40.0ml乙醇中,加入水合肼5.0g,搅拌反应混合物并加热至回流过夜;通过tlc监测反应完毕后,减压除去溶剂,析出的白色晶体用乙醇重结晶,得到4.43g化合物(v),收率82%。
将化合物(ii)5.83g溶于50.0ml乙醇中,加入2.51g化合物(v),搅拌反应混合物并加热至回流36小时,然后冷却至室温。用乙酸乙酯进行萃取,有机层分别用水10.0ml洗涤3次。通过柱色谱用甲醇/乙酸乙酯(1:30)纯化,并真空干燥。得到化合物(vi)3.82g,收率90%。
将化合物(vi)3.82g溶于40.0ml乙醇中,加入水合肼1.63g,搅拌反应混合物并加热至回流过夜;通过tlc监测反应完毕后,减压除去溶剂,析出的白色晶体用乙醇重结晶,得到2.94g化合物(b),收率82%。
步骤3:化合物(c)3-甲酰基-7-(二乙氨基)香豆素席夫碱的制备
将化合物(a)1.72g溶于40.0ml乙醇中,加入化合物(b)1.53g,搅拌反应混合物并加热至回流过夜,通过tlc监测反应完毕后,减压除去溶剂,析出的橙红色晶体用乙醇重结晶,得到2.49g化合物(c)3-甲酰基-7-(二乙氨基)香豆素席夫碱,收率80%。
实施例2
步骤1:化合物a的制备
将4-(二乙基氨基)水杨醛(1)1.85g溶于40.0ml乙醇中,加入丙二酸二乙酯(2)3.12ml和哌啶1ml,加热搅拌至回流过夜;其后,通过tlc监测反应完毕后,减压除去产物中的溶剂乙醇,然后加入浓盐酸20.0ml和冰醋酸20.0ml进行水解并再搅拌6小时;冷却后倒入100ml冰水中,再加入40wt%naoh水溶液调节溶液的ph至7;其后向溶液中加入30.0ml二氯甲烷进行萃取,分别用水10.0ml洗涤萃取有机层3次后,再经无水mgso4干燥、真空浓缩、柱色谱法用乙酸乙酯/己烷(1∶3)纯化,得到1.95g4-(二乙氨基)香豆素,收率93.7%;
在50℃、ar保护下,将干燥的dmf2.14ml滴加到pocl32.18ml中并搅拌30分钟,得到红色溶液;将4-(二乙氨基)香豆素1.95g溶于10.0mldmf中,并通过恒压滴液漏斗缓慢滴加到红色溶液中,在70℃下搅拌反应12小时,然后将产物倒入100ml冰水中,加入20wt%naoh水溶液调节溶液的ph至7;其后用30.0ml乙酸乙酯分批萃取,合并有机相后用饱和食盐水洗涤3次,再经用无水mgso4干燥、真空浓缩、柱色谱法用乙酸乙酯/己烷(1∶2)纯化,得到化合物(a)1.7g,收率77%。
步骤2:化合物(b)的制备
将干燥的氯化氢用鼓泡法溶于无水乙醇溶剂中,滴加5.2g2-氰基乙酸乙酯。在0℃下反应4小时后有白色沉淀析出。减压旋蒸除去溶剂乙醇,析出的白色晶体用石油醚重结晶,得到化合物(ii)6.14g。收率84%。
将化合物(iii)6.15g溶于50.0ml无水乙醇中,滴加2滴浓硫酸,搅拌反应混合物并加热至回流4小时,通过tlc监测反应完毕后,减压除去溶剂,再通过柱色谱用乙酸乙酯/正己烷(1:3)纯化,得到化合物(iv)6.04g,收率80%。
将化合物(iv)6.04g溶于40.0ml无水乙醇中,加入水合肼5.0g,搅拌反应混合物并加热至回流过夜;通过tlc监测反应完毕后,减压除去溶剂,析出的白色晶体用无水乙醇重结晶,得到4.49g化合物(v),收率82%。
将化合物(ii)6.14g溶于50.0ml无水乙醇中,加入2.645g化合物(v),搅拌反应混合物并加热至回流36小时,然后冷却至室温。用乙酸乙酯进行萃取,有机层分别用水10.0ml洗涤3次。通过柱色谱用甲醇/乙酸乙酯(1:30)纯化,并真空干燥。得到化合物(vi)3.98g,收率89%。
将化合物(vi)3.98g溶于40.0ml乙醇中,加入水合肼1.7g,搅拌反应混合物并加热至回流过夜;通过tlc监测反应完毕后,减压除去溶剂乙醇,析出的白色晶体用无水乙醇重结晶,得到3.17g化合物(b),收率85%。
步骤3:化合物(c)3-甲酰基-7-(二乙氨基)香豆素席夫碱的制备
将化合物(a)1.7g溶于40.0ml无水乙醇中,加入化合物(b)1.51g,搅拌反应混合物并加热至回流过夜,通过tlc监测反应完毕后,减压除去溶剂,析出的橙红色晶体用无水乙醇重结晶,得到2.5g化合物(c)3-甲酰基-7-(二乙氨基)香豆素席夫碱,收率81%。
- 还没有人留言评论。精彩留言会获得点赞!