吗啉基喹唑啉类化合物的晶型、其制备方法及应用与流程
本发明涉及吗啉基喹唑啉类化合物的的晶型、其制备方法及应用。
背景技术:
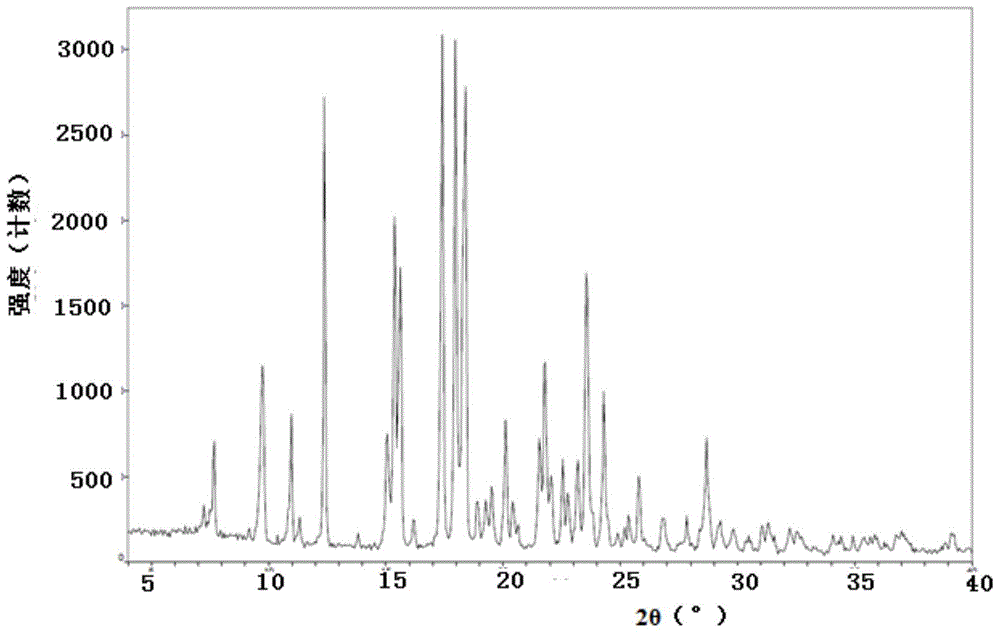
:一种吗啉基喹唑啉类化合物,其结构如式a所示(以下简称如式a所示的吗啉基喹唑啉类化合物),具有抑制磷脂酰肌醇3-激酶δ(pi3kδ)的活性。pi3kδ是一种胞内磷酸酰肌醇激酶,可催化磷脂酰醇的3位羟基磷酸化。pi3k可分为i类、ii类和iii类激酶,而研究最广泛的是能被细胞表面受体所激活的i类pi3k。哺乳动物细胞中i类pi3k根据结构和受体又分为ia类和ib类,它们分别传递来自酪氨酸激酶-偶联受体和g蛋白-偶联受体的信号。ia类pi3k包括pi3kα、pi3kβ、pi3kδ亚型,ib类pi3k包括pi3kγ亚型(trends.biochem.sci.,1997,22,267-272)。ia类pi3k是由催化亚单位p110和调节亚单位p85所组成的二聚体蛋白,具有类脂激酶和蛋白激酶的双重活性(nat.rev.cancer2002,2,489-501),被认为与细胞增殖和癌症发生,免疫疾病和涉及炎症的疾病相关。专利wo2015055071a1公开了如式a所示的吗啉基喹唑啉类化合物及其制备方法。如式a所示的吗啉基喹唑啉类化合物的晶型对药物在生产、加工、储存、运输时的稳定性有至关重要的影响。物质可以以两种或两种以上不同的晶体结构存在的现象称为多晶型现象。而化合物不同的固体形式往往表现出不同的物理和化学性质。对于药物而言,这种多晶型现象可能会影响到药物的吸收,进而影响药物的生物利用度,从而表现出不同的临床疗效和毒副作用。鉴于此,开发具有优势性能的如式a所示的吗啉基喹唑啉类化合物的晶型具有十分重要的意义。技术实现要素:本发明所要解决的技术问题是提供一种如式a所示的吗啉基喹唑啉类化合物的晶型、制备方法及应用,该晶型制备方法简单,适合工业化生产,且不易吸湿,具有较好的稳定性,对药物的优化和开发具有重要的价值。本发明通过以下技术方案解决上述技术问题。本发明提供了一种如式a所示的吗啉基喹唑啉类化合物的晶型i,其以2θ角表示的x-射线粉末衍射图,在7.7±0.2°、9.7±0.2°、12.4±0.2°、15.4±0.2°、17.4±0.2°、18.0±0.2°和18.4±0.2°处有衍射峰;所述如式a所示的吗啉基喹唑啉类化合物的晶型i,其以2θ角表示的x-射线粉末衍射图,还可在如下一个或多个2θ角处有衍射峰:11.0±0.2°、11.3±0.2°、19.5±0.2°、20.1±0.2°、21.8±0.2°、22.6±0.2°、23.2±0.2°、23.6±0.2°、24.3±0.2°、25.8±0.2°和28.7±0.2°。所述如式a所示的吗啉基喹唑啉类化合物的晶型i,其以2θ角表示的x-射线粉末衍射图,可在7.7±0.2°、9.7±0.2°、11.0±0.2°、12.4±0.2°、15.4±0.2°、17.4±0.2°、18.0±0.2°、18.4±0.2°、23.6±0.2°和24.3±0.2°处有衍射峰。较佳地,所述如式a所示的吗啉基喹唑啉类化合物的晶型i,其以2θ角表示的x-射线粉末衍射图,在7.7±0.2°、9.7±0.2°、11.0±0.2°、11.3±0.2°、12.4±0.2°、15.4±0.2°、17.4±0.2°、18.0±0.2°、18.4±0.2°、19.5±0.2°、20.1±0.2°、21.8±0.2°、22.6±0.2°、23.2±0.2°、23.6±0.2°、24.3±0.2°、25.8±0.2°和28.7±0.2°处有衍射峰。本发明中,所述如式a所示的吗啉基喹唑啉类化合物的晶型i,其以2θ角表示的x-射线粉末衍射图,其衍射峰和峰高百分比还可如表1所示:表1编号2θ(±0.2°)峰高百分比(%)17.2395.527.66618.439.73234.5410.96225.7511.3185.4612.38589.2715.37765.5817.404100.0917.97199.41018.38289.61119.51611.01220.11124.61321.79536.01422.55115.81523.19116.51623.56453.21724.30030.51825.79913.91928.68421.5本发明中,所述如式a所示的吗啉基喹唑啉类化合物的晶型i,其以2θ角表示的x-射线粉末衍射图,其衍射峰和峰面积百分比还可如表2所示:表2编号2θ(±0.2°)峰面积百分比(%)17.2396.427.66615.539.73237.4410.96218.9511.3184.3612.38552.4715.37764.0817.40476.1917.97187.31018.382100.01119.51611.11220.11120.61321.79543.61422.55111.11523.19118.11623.56460.91724.30026.61825.79914.51928.68424.2本发明中,所述如式a所示的吗啉基喹唑啉类化合物的晶型i,其以2θ角度表示的x-射线粉末衍射图,其衍射峰、峰宽值、峰高百分比和峰面积百分比还可如表3所示:表3本发明中,所述如式a所示的吗啉基喹唑啉类化合物的晶型i以2θ角表示的x-射线粉末衍射图还可基本上如图1所示。本发明中,所述的x-射线粉末衍射图均使用cu靶的kα谱线测得。本发明中,所述如式a所示的吗啉基喹唑啉类化合物的晶型i的红外吸收光谱图(ir)还可在以下位置处有特征峰:3445cm-1、3246cm-1、3018cm-1、3001cm-1、2972cm-1、2953cm-1、2924cm-1、2910cm-1、2891cm-1、2850cm-1、1604cm-1、1589cm-1、1552cm-1、1506cm-1、1489cm-1、1458cm-1、1413cm-1、1365cm-1、1155cm-1和775cm-1。本发明中,所述如式a所示的吗啉基喹唑啉类化合物的晶型i的红外吸收光谱图中的特征峰、振动类型、基团和吸收峰强度还可如表4所示:表4本发明中,所述如式a所示的吗啉基喹唑啉类化合物的晶型i的红外吸收光谱图还可基本上如图2所所示。本发明中,所述如式a所示的吗啉基喹唑啉类化合物的晶型i的热重分析图(tga)还可基本上如图3所示。本发明中,所述如式a所示的吗啉基喹唑啉类化合物的晶型i的差示扫描量热图(dsc)中还可在204.3±3℃处有吸收峰,熔化热为98.70j/g。本发明中,所述如式a所示的吗啉基喹唑啉类化合物的晶型i的差示扫描量热图还可基本上如图4所示。本发明中,所述如式a所示的吗啉基喹唑啉类化合物的晶型i的动态水分吸附图(dvs)中,所述晶型i增加的质量相比于初始的质量,还可在0%~90%相对湿度范围内增重了0.23%,在0%~95%相对湿度范围内所述晶型i的质量增重了0.34%。本发明中,所述如式a所示的吗啉基喹唑啉类化合物的晶型i的动态水分吸附图还可基本上如图5所示。本发明还提供了一种如式a所示的吗啉基喹唑啉类化合物的晶型i的制备方法;其为方法1或方法2;方法1:将式a所示的吗啉基喹唑啉类化合物在溶剂中形成热饱和溶液,冷却,即可;所述溶剂选自乙腈、2-甲基四氢呋喃、丙酮、乙酸乙酯、乙醇和异丙醇中的一种或多种。所述热饱和溶液的配置方法可参考本领域的常规配置方法,较佳地,在所述的冷却步骤前,所述热饱和溶液经过滤处理。所述过滤处理的方式可为本领域此类操作常规的过滤方式,较佳地为热过滤。所述热过滤为滤膜过滤。所述滤膜的孔径较佳地为0.45微米。所述冷却的方式可为本领域此类操作的常规所用的冷却方式,较佳地为快速冷却的方式或缓慢冷却的方式。较佳地,当所述冷却的方式为快速冷却时,所述冷却的最终温度为-15~-25℃,例如-20℃。较佳地,当所述冷却的方式为缓慢冷却时,所述冷却的速率为5~15℃/h,例如10℃/h。本发明中,所述冷却后还可包括后处理步骤:过滤和干燥。所述过滤可为本领域此类操作的常规条件和操作,较佳地为减压过滤。所述干燥可为本领域此类操作的常规条件和操作,较佳地为真空干燥。方法2:将如式a所示的吗啉基喹唑啉类化合物在溶剂a和溶剂b中混合,溶解,析晶,即可;所述溶剂a选自四氢呋喃、1,4-二氧六环、乙醇、乙酸乙酯、n,n-二甲基甲酰胺(dmf)、n,n-二甲基乙酰胺(dmac)和二甲基亚砜(dmso)中的一种或多种;所述溶剂b选自正庚烷、正己烷、环己烷、环戊烷、正戊烷、石油醚和水中的一种或多种。较佳地,当所述溶剂a选自四氢呋喃、1,4-二氧六环、乙醇和乙酸乙酯中的一种或多种时,所述溶剂b选自正庚烷、正己烷、环己烷、环戊烷、正戊烷和石油醚中的一种或多种。较佳地,当所述溶剂a选自n,n-二甲基甲酰胺(dmf)、n,n-二甲基乙酰胺(dmac)和二甲基亚砜(dmso)中的一种或多种,所述溶剂b为水。所述析晶的方式可为本领域此类操作的常规方式。较佳地为自然冷却至室温析晶的方式。所述溶解的条件,可为本领域此类操作的常规条件。较佳地,所述溶解的条件为加热;更佳为加热并搅拌。所述加热的温度一般为溶解如式a所示的吗啉基喹唑啉类化合物的溶剂的沸点,较佳地为40~90℃,例如50℃。所述搅拌的转速可为200~350rpm,例如260rpm。较佳地,所述溶解之后还可包括过滤步骤。所述过滤可为本领域此类操作常规的过滤方式,较佳地为滤膜过滤。所述滤膜的孔径较佳地为0.45微米。所述方法2进一步优选以下步骤:将所述如式a所示的吗啉基喹唑啉类化合物溶解在所述溶剂a中,得到混合溶液,将所述溶剂b加入至所述混合溶液中,析晶,即可。较佳地,所述加入的方式可为滴加的方式。本发明中,所述析晶之后还可包括以下后处理步骤:过滤和干燥。所述过滤可为本领域此类操作的常规条件和操作,较佳地为减压过滤。所述干燥可为本领域此类操作的常规条件和操作,较佳地为真空干燥。本发明还提供了一种所述如式a所示的吗啉基喹唑啉类化合物的晶型i在制备激酶抑制剂中的应用。其中,所述的所述激酶优选为pi3激酶(pi3k),还可以优选为pi3激酶(pi3k)的p110δ亚型。本发明还提供了一种所述如式a所示的吗啉基喹唑啉类化合物的晶型i在制备药物中的应用,所述的药物用于防治和/或治疗与激酶有关的疾病。本发明中,所述激酶优选为pi3激酶(pi3k),进一步优选pi3激酶(pi3k)的p110δ亚型。本发明中,所述的与激酶有关的疾病包括但不限于:癌症、免疫疾病、代谢和/或内分泌功能障碍、心血管疾病、病毒感染和炎症、和神经疾病中的一种或多种,优选癌症和/或免疫疾病。其中,所述的免疫疾病包括但不限于类风湿性关节炎、牛皮癣、溃疡性结肠炎、克罗恩病和全身性红斑狼疮中的一种或多种。所述的心血管疾病包括但不限于血液肿瘤。所述的病毒感染和炎症包括但不限于哮喘和/或特异性皮炎。本发明还提供了一种所述如式a所示的吗啉基喹唑啉类化合物的晶型i在制备药物中的应用,所述的药物用于防治和/或治疗疾病,所述的疾病为癌症、免疫疾病、代谢和/或内分泌功能障碍、心血管疾病、病毒感染、炎症和神经疾病中的一种或多种。其中,所述的免疫疾病、所述的心血管疾病、所述的病毒感染和炎症均同上所述。本发明还提供了一种药物组合物,其包含所述如式a所示的吗啉基喹唑啉类化合物的晶型i,和药学上可接受的载体。本发明还提供了一种上述药物组合物在制备用于防治和/或治疗与激酶有关的疾病的药物中的应用。其中,所述的与激酶有关的疾病同前所述。本发明还提供了一种上述药物组合物在制备用于防治和/或治疗“癌症、免疫疾病、代谢和/或内分泌功能障碍、心血管疾病、病毒感染和炎症,和神经疾病中的一种或多种”的药物中的应用。其中,所述的免疫疾病、所述的心血管疾病、所述的病毒感染和炎症均同上所述。本发明中,所述如式a所示的吗啉基喹唑啉类化合物的晶型i也可以与一种或多种其他活性成分组合使用;当组合使用时,活性成分可以是分开的组合物,用于在治疗中通过相同或不同的施用途径同时施用或者在不同时间分别施用,或者它们也可以在同一药物组合物中一起施用。本发明中,所述药物组合物的给药方法没有特殊限制,可根据病人年龄、性别和其它条件及症状,选择各种剂型的制剂给药;例如,片剂、丸剂、溶液、悬浮液、乳液、颗粒剂或胶囊口服给药;针剂可以单独给药,或者和注射用输送液(如葡萄糖溶液及氨基酸溶液)混合进行静脉注射;栓剂为给药到直肠。在一些实例中,所述如式a所示的吗啉基喹唑啉类化合物的晶型i不会在与一种或多种药学上可接受的载体和/或辅料和/或稀释剂制成制剂时发生转化。本发明中,“防治”指“预防”。“预防”是指获得或发生疾病或障碍的风险降低(即导致可能暴露于导致疾病试剂或疾病发作前易感疾病的受试者中未发生疾病的临床症状的至少一种)。本发明中,“治疗”指改善疾病或障碍(即阻止疾病或减少表现、其临床症状的程度或严重性);或者,改善至少一种身体参数,其可能不被受试者察觉;或者减缓疾病进展。本发明的晶型可以通过一种或几种固态分析方法进行鉴定。如x射线粉末衍射、单晶x-射线衍射、红外吸光光谱、差示扫描量热、热重曲线等。本领域技术人员知道,x射线粉末衍射的峰强度和/或峰情况可能会因为实验条件不同而不同。同时由于仪器不同的精确度,测得的2θ值会有约±0.2°的误差。而峰的相对强度值比峰的位置更依赖于所测定样品的某些性质,如晶体的尺寸大小,纯度高低,因此测得的峰强度可能出现约±20%的偏差。尽管存在试验误差、仪器误差和取向优先等,本领域技术人员还是可以从本专利提供的x射线粉末衍射数据获得足够的鉴别各个晶型的信息。在红外光谱测定中,由于各种型号的仪器性能不同、供试品制备时研磨程度的差异或吸水程度不同等原因,对光谱的形状及吸收峰的位置均会有一定程度的影响。而在dsc测量中,根据加热速率、晶体形状和纯度和其它测量参数,实测获得的吸热峰的初始温度、最高温度和熔化热数据均具有一定程度的可变性。本发明中,术语“快速冷却”是指将饱和热溶液直接放置在远低于该饱和溶液中的溶剂的沸点的温度下(例如-20℃),进行冷却,该过程冷却速度快。本发明中,术语“缓慢冷却”是指饱和热溶液以5~15℃/h的速率(例如10℃/h)冷却至室温,该过程冷却速度慢。本发明中,“室温”是指“10~30℃”。在不违背本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。本发明所用试剂和原料均市售可得。本发明的积极进步效果在于:本发明提供的如式a所示的吗啉基喹唑啉类化合物晶型i制备方法简单,且具有较好的稳定性,且不易吸湿,对药物的优化和开发具有重要的价值。附图说明图1为实施例1得到如式a所示的吗啉基喹唑啉类化合物的晶型i的x-射线粉末衍射图。图2为实施例1得到如式a所示的吗啉基喹唑啉类化合物的晶型i的红外吸收光谱图。图3为实施例1得到如式a所示的吗啉基喹唑啉类化合物的晶型i的热重分析图。图4为实施例1得到如式a所示的吗啉基喹唑啉类化合物的晶型i的差示扫描量热图。图5为实施例1得到如式a所示的吗啉基喹唑啉类化合物的晶型i的动态水分吸附图;其中1为去湿曲线,2为吸湿曲线。图6为对比实施例1得到如式a所示的吗啉基喹唑啉类化合物的晶型ii的x-射线粉末衍射图。图7为对比实施例1得到如式a所示的吗啉基喹唑啉类化合物的晶型ii的热重分析图。图8为对比实施例1得到如式a所示的吗啉基喹唑啉类化合物的晶型ii的差示扫描量热图。图9为对比实施例1得到如式a所示的吗啉基喹唑啉类化合物的晶型ii的动态水分吸附图;其中1为去湿曲线,2为吸湿曲线。图10为根据专利wo2015055071a1方法得到如式a所示的吗啉基喹唑啉类化合物的无定型的x-射线粉末衍射图。具体实施方式下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。检测方法仪器粉末x-射线衍射分析(xrpd):光源为cuk,x-射线强度为40kv/40ma,扫描模式为theta-theta,扫描角度范围4°~40°,步长为0.05°,扫描速度为0.5秒/步。红外吸收光谱分析(ir):依据中国药典2015年版四部通则0402红外分光光度法,采用溴化钾压片法制备供试品,在4000~400cm-1波数范围内采集红外吸收光谱。供试品的扫描次数为45次,仪器分辨率为4cm-1。差示扫描量热分析(dsc):称取样品2~4mg,置于非密闭铝盘中,氮气流(50ml/min)环境中,样品在25℃平衡,然后以10℃/min的升温速率从25℃加热至300或400℃。热失重分析(tga):称取样品8~12mg,置于铂金样品盘中,氮气流(50ml/min)环境中,以10℃/min的升温速率从25℃加热至300或400℃。动态水分吸附分析(dvs):取样品约10mg,温度设定25℃,湿度0%rh条件下干燥60分钟后,测试湿度从0%rh~95%rh变化时样品的吸湿特征,以及湿度从95%rh~0%rh变化时样品的去湿特征,湿度变化步长5%rh,当质量变化率dm/dt的值小于0.002%时视为天平平衡,当5分钟内质量变化率小于0.01%/分钟为检测过程中的平衡标准,最长平衡时间为2小时。测定该测试条件下的等温吸附/脱附水的特征,并对dvs测试后的样品进行xrpd检测。根据专利wo2015055071a1中实施例10的合成方法,制备得到如式a所示的吗啉基喹唑啉类化合物,经xrpd表征为无定型,其xrpd图如图10所示,其效果数据如表9所示。实施例1如式a所示的吗啉基喹唑啉类化合物的晶型i的制备称取约20mg如式a所示的吗啉基喹唑啉类化合物到小瓶中,往小瓶中加入一定体积的乙腈,超声2分钟,将样品瓶置于磁力加热搅拌器上,控制温度为50℃,转速为260rpm,加热促进样品溶解,若溶液已澄清,则添加一定量的固体样品,继续加热促溶,保证最后得到样品过饱和溶液,趁热用0.45微米滤膜过滤并转移到新的小瓶中。将小瓶立即放到-20℃冰箱中,析出固体过滤得到样品。经x-射线粉末衍射图谱测定所得样品为晶型i。x-射线粉末衍射图如图1所示,其以2θ角表示的x-射线粉末衍射,在7.7±0.2°,9.7±0.2°,11.0±0.2°,11.3±0.2°,12.4±0.2°,15.4±0.2°,17.4±0.2°,18.0±0.2°,18.4±0.2°,19.5±0.2°,20.1±0.2°,21.8±0.2°,22.6±0.2°,23.2±0.2°,23.6±0.2°,24.3±0.2°,25.8±0.2°和28.7±0.2°处有衍射峰;其ir图如图2所示,在3445cm-1,3246cm-1,3018cm-1,3001cm-1,2972cm-1,2953cm-1,2924cm-1,2910cm-1,2891cm-1,2850cm-1,1604cm-1,1589cm-1,1552cm-1,1506cm-1,1489cm-1,1458cm-1,1413cm-1,1365cm-1,1155cm-1,775cm-1处有特征峰。tga图如图3所示。由图3可知,晶型i为无水物,不含水或溶剂。其dsc图如图4所示。晶型i的差示扫描量热中在204.3±3℃处有吸收峰,熔化热为98.70j/g。其dvs图如图5所示。晶型i的态水分吸附图中,所述晶型i增加的质量相比于初始的质量,在0%~90%相对湿度范围内增重0.23%,在0%~95%相对湿度范围内增重0.34%。实施例2如式a所示的吗啉基喹唑啉类化合物的晶型i的制备称取约20mg如式a所示的吗啉基喹唑啉类化合物到小瓶中,往小瓶中加入一定体积的乙腈,超声2分钟,将样品瓶置于磁力加热搅拌器上,控制温度为50℃,转速为260rpm,加热促进样品溶解,若溶液已澄清,则添加一定量的固体样品,继续加热促溶,保证最后得到样品过饱和溶液,趁热用0.45微米滤膜过滤并转移到新的小瓶中。以10℃/h的速率缓慢降温至室温(25℃)并在室温下搅拌过夜,析出固体过滤得到样品。此方法所得样品的x射线粉末衍射图谱经与实施例1样品图谱比对,确定为晶型i。实施例3如式a所示的吗啉基喹唑啉类化合物的晶型i的制备方法同实施例2,将溶剂改为2-甲基四氢呋喃、丙酮、乙酸乙酯、乙醇、异丙醇,析出固体过滤得到样品。此方法所得样品的x射线粉末衍射图谱经与实施例1样品图谱比对,确定为晶型i。实施例4如式a所示的吗啉基喹唑啉类化合物的晶型i的制备称取约1g如式a所示的吗啉基喹唑啉类化合物,加5ml丙酮,在加热下溶解,停止加热,放置过夜。第二天,过滤,干燥得到样品。此方法所得样品的x射线粉末衍射图谱经与实施例1样品图谱比对,确定为晶型i。实施例5如式a所示的吗啉基喹唑啉类化合物的晶型i的制备称取约20mg如式a所示的吗啉基喹唑啉类化合物到小瓶中,往小瓶中加入一定体积的四氢呋喃,超声2分钟,将样品瓶置于磁力加热搅拌器上,控制温度为50℃,转速为260rpm,加热促进样品溶解,若溶液已澄清,则添加一定量的固体样品,继续加热促溶,保证最后得到样品呈过饱和溶液,趁热用0.45微米滤膜过滤并转移到新的小瓶中,向瓶中缓慢滴加10倍体积的正庚烷并保持缓慢搅拌,析出固体过滤得到样品。此方法所得样品的x射线粉末衍射图谱经与实施例1样品图谱比对,确定为晶型i。实施例6如式a所示的吗啉基喹唑啉类化合物的晶型i的制备方法同实施例5,将溶剂四氢呋喃改为将二氧六环,不良溶剂为正庚烷,滴加的体积倍数为13倍。此方法所得样品的x射线粉末衍射图谱经与实施例1样品图谱比对,确定为晶型i。实施例7如式a所示的吗啉基喹唑啉类化合物的晶型i的制备在室温下,将1g如式a所示的吗啉基喹唑啉类化合物溶解于5.5ml的dmf中,搅拌下慢慢加入1ml水,析出固体过滤得到样品。此方法所得样品的x射线粉末衍射图谱经与实施例1样品图谱比对,确定为晶型i。实施例8如式a所示的吗啉基喹唑啉类化合物的晶型i的制备往10g如式a所示的吗啉基喹唑啉类化合物中加入500ml乙醇,在加热下完全溶解,趁热过滤,滤液浓缩至50-70ml后,室温搅拌过夜,加入正庚烷析晶到大量固体析出,过滤,在小于85℃下真空干燥5-6小时,得到样品。此方法所得样品的x射线粉末衍射图谱经与实施例1样品图谱比对,确定为晶型i。实施例9如式a所示的吗啉基喹唑啉类化合物的晶型i的制备往5g如式a所示的吗啉基喹唑啉类化合物中加入25ml乙醇和25ml正庚烷,回流16小时后,冷却至室温,过滤,小于85℃下真空干燥16小时,得到约4g样品。此方法所得样品的x射线粉末衍射图谱经与实施例1样品图谱比对,确定为晶型i。实施例10如式a所示的吗啉基喹唑啉类化合物的晶型i的制备在室温下,往5g如式a所示的吗啉基喹唑啉类化合物中加入5.5ml的dmso溶解,慢慢加入5ml水析出固体,过滤,小于85℃下真空干燥17小时,得到样品。此方法所得样品的x射线粉末衍射图谱经与实施例1样品图谱比对,确定为晶型i。实施例11如式a所示的吗啉基喹唑啉类化合物的晶型i的制备在室温下,往6g如式a所示的吗啉基喹唑啉类化合物中加入乙酸乙酯360g溶解,浓缩到一半体积,加入约60g正庚烷,析出固体,过滤。小于85℃下真空干燥48小时,得到样品。此方法所得样品的x射线粉末衍射图谱经与实施例1样品图谱比对,确定为晶型i。对比实施例1如式a所示的吗啉基喹唑啉类化合物的晶型ii的制备称取30mg如式a所示的吗啉基喹唑啉类化合物到小瓶中,往小瓶中加入1,4-二氧六环/异丙醚(v/v=1:1),超声促使样品溶解,若溶液已澄清,则继续添加一定量的固体样品,并超声促溶,保证最后得到样品为过饱和溶液,用0.45微米滤膜过滤并转移到新的小瓶,将小瓶敞口放置,室温下自然挥发溶剂,得到固体即为如式a所示的吗啉基喹唑啉类化合物的晶型ii。x-射线粉末衍射图谱如图6所示,其中2θ、d(a)、峰高数和峰面积如下表5所示:表5tga图如图7所示。由图7可知,如式a所示的吗啉基喹唑啉类化合物晶型ii为无水物。dsc图如图8所示。如式a所示的吗啉基喹唑啉类化合物晶型ii的差示扫描量热中在202.83±3℃处有吸收峰,熔化热为83.42j/g。dvs图如图9所示。如式a所示的吗啉基喹唑啉类化合物晶型ii的动态水分吸附图中,在0%~95%相对湿度范围内增重6.237%。效果实施例1稳定性1如式a所示的吗啉基喹唑啉类化合物的晶型i在水和有机溶剂中的稳定性1.1如式a所示的吗啉基喹唑啉类化合物的晶型i在水和有机溶剂中室温放置10天的稳定性分别称取约20mg如式a所示的吗啉基喹唑啉类化合物晶型i固体样品到小瓶子,分别往小瓶中加入1ml的水或有机溶剂,超声5分钟得到混悬液。将混悬液在室温下旋转10天,过滤后的湿样品用xrpd表征。结果显示,各种溶剂下,晶型都没有发生变化,仍为晶型i。所述的溶剂为水、甲醇,乙醇,乙酸乙酯,丙酮,甲基叔丁基醚,乙腈,正己烷,异丙醇,正庚烷,甲苯,甲乙酮,异丙醚,乙酸异丙酯,正丁醇,甲醇的水溶液(90%,75%,50%,10%),丙酮的水溶液(95%,85%,15%)。1.2如式a所示的吗啉基喹唑啉类化合物的晶型i在有机溶剂中高温打浆18小时的稳定性往5g如式a所示的吗啉基喹唑啉类化合物晶型i固体中加入乙醇10g,异丙醇10g,正庚烷10g。混合物在80℃打浆18小时,冷却至室温,过滤,小于85℃真空干燥16小时,得到约4g样品。此方法所得样品的x射线粉末衍射图谱与实施例1中所得晶型i样品的衍射峰一致。如式a所示的吗啉基喹唑啉类化合物的晶型i在水和有机溶剂中经过长时间的室温放置,以及高温放置均没有发生变化,可见,其在水和有机溶剂中较好的稳定性。2如式a所示的吗啉基喹唑啉类化合物的晶型i在高温、高湿、光照条件下的稳定性将适量的如式a所示的吗啉基喹唑啉类化合物的晶型i的样品置于培养皿上,分别于高温(40±2℃和60±2℃)、高湿(25℃,rh75±5%和rh90±5%)和光照(4500±500lux,25℃)条件下敞开放置。于5天、10天、1月取样试验,结果见下表6~8:将适量的如式a所示的吗啉基喹唑啉类化合物无定型样品和如式a所示的吗啉基喹唑啉类化合物晶型ii的样品分别置于培养皿上,分别于高温(60±2℃)、高湿(25℃,rh75±5%)和光照(4500±500lux,25℃)条件下敞开放置。于5天、10天取样试验,结果见下表9~10:表6影响因素高温(40±2℃、60±2℃)试验结果表7影响因素高湿(25℃,rh75±5%、rh90±5%)试验结果表8影响因素光照(4500lux±500lux,25℃)试验结果表9影响因素高温(60±2℃)、光照(4500lux±500lux,25℃)、高湿(25℃,rh75±5%)试验结果表10影响因素高温(60±2℃)、光照(4500lux±500lux,25℃)、高湿(25℃,rh75±5%)试验结果由上述表6~8数据表明,如式a所示的吗啉基喹唑啉类化合物的晶型i在高温、高湿、光照条件下,其化学纯度和晶型均没有发生改变,具有较好的稳定性。如式a所示的吗啉基喹唑啉类化合物的无定型样品分别在光照(4500±500lux,25℃)、高温(60℃)和高湿(25℃,rh75%)条件下放置10天,性状无明显变化。由上述表9数据表明,样品在高温(60℃)和高湿(25℃,rh75%)条件下总杂含量略有增加;在光照(4500±500lux,25℃)条件下,总杂含量显著增大,样品在光照条件下不稳定。获得如式a所示的吗啉基喹唑啉类化合物的晶型ii难度较大,纯度也稍差。样品分别在光照(4500±500lux,25℃)、高温(60℃)和高湿(25℃,rh75%)条件下放置10天,性状无明显变化。由上述表10数据表明,样品在高温(60℃)和高湿(rh75%)条件下总杂含量略有增加;在光照(4500±500lux)条件下,总杂含量显著增大,样品在光照条件下不稳定。可见,如式a所示的吗啉基喹唑啉类化合物的晶型i在高温、高湿、光照条件下,均具有较好的稳定性。效果实施例2吸湿性取样品约10mg,温度设定25℃,湿度0%rh条件下干燥60分钟后,测试湿度从0%rh~95%rh变化时样品的吸湿特征,以及湿度从95%rh~0%rh变化时样品的去湿特征,湿度变化步长5%rh,当质量变化率dm/dt的值小于0.002%时视为天平平衡,当5分钟内质量变化率小于0.01%/分钟为检测过程中的平衡标准,最长平衡时间为2小时。测定该测试条件下的等温吸附/脱附水的特征,并对dvs测试后的样品进行xrpd检测。由图5如式a所示的吗啉基喹唑啉类化合物的晶型i的dvs可知,所述晶型i增加的质量相比于初始的质量,在0~90%相对湿度范围内增重0.23%,在0%~95%相对湿度范围内增重0.34%。由图9如式a所示的吗啉基喹唑啉类化合物的晶型ii的dvs可知,所述如式a所示的吗啉基喹唑啉类化合物晶型ii增加的质量相比于初始的质量,在0~95%相对湿度范围内增重6.237%。在0~95%相对湿度范围内如式a所示的吗啉基喹唑啉类化合物晶型ii增重量为如式a所示的吗啉基喹唑啉类化合物晶型i的18倍,可见如式a所示的吗啉基喹唑啉类化合物晶型i具有较小的吸湿性。可见,本发明的如式a所示的吗啉基喹唑啉类化合物的晶型i具有较好的稳定性和极低的吸湿性。应当理解,本文所述的实施例仅用于说明目的,通过实施例将有助于进一步理解本发明,但不用于限制本发明的内容。对于本领域技术人员而言,对于材料和方法两者的许多改变可在不脱离本发明范围的情况下实施,这些改变或改进包括在本申请的主旨和范围以及所附权利要求的范围内。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1