一种羟肟酸类捕收剂的合成方法与流程
本发明涉及捕收剂合成技术领域,具体涉及一种羟肟酸类捕收剂的合成方法。
背景技术:
羟肟酸是一类典型金属离子螯合剂,分子结构如下:
因其特殊的分子结构,对不同的金属矿物具有良好的选择性捕收性能,在钨、锡、稀土等矿物的浮选中具有广泛的应用。
目前,工业上一般采用“羟胺法”制备羟肟酸捕收剂,即酯与羟胺在碱性条件下进行羟肟化反应制备羟肟酸。现有工艺存在转化率低、产品质量不稳定等技术问题。主要包括以下原因:一方面,为了避免固体产品溶于大量水中造成损失,现有工艺尽量降低溶剂水的用量,这导致反应过程或酸化过程结块严重,物料的混合不充分,反应不能进行的较为彻底;另一方面,反应物酯与羟胺分属油、水两相,单靠机械搅拌不易达到充分的接触,反应速率较慢、转化率低。
针对以上问题,有必要对羟肟酸类捕收剂的合成进行研究,以期得到一种新的合成羟肟酸捕收剂的方法,来有效解决以上问题。
技术实现要素:
本发明解决的技术问题是提供一种羟肟酸类捕收剂的合成方法,有效提高反应转化率。
为解决上述技术问题,本发明采用以下技术方案:一种羟肟酸类捕收剂的合成方法,所述合成方法的反应式为:
式(1)所示的反应物酯与式(2)所示的羟胺盐在催化剂dbu、碱moh的作用下,反应得到中间产物式(3)所示化合物,式(3)所示化合物再进行酸解得到式(4)所示的羟肟酸类捕收剂:
其中,r1为芳基、环烷基、取代的芳基或取代的环烷基,所述取代基为卤素、c1-6的烷基、c3-6的环烷基、苯基中的任意一种;
所述式(2)所示的羟胺盐为羟胺盐酸盐或羟胺硫酸盐;
所述的碱moh为氢氧化钠或氢氧化钾,m为k或na。
作为一种优选的实施方案,羟肟酸类捕收剂的合成方法,包括以下步骤:
(1)将式(1)所示的反应物酯与催化剂dbu混合,之后搅拌5min~1h,得到混合物1;
(2)将式(2)所示的羟胺盐、碱moh分别加水配制成质量百分比浓度为20~50%的羟胺盐水溶液和碱溶液;
(3)将所述混合物1加入到所述羟胺盐水溶液中,得到混合液a,之后在搅拌条件下向所述混合液a中分批加入所述碱溶液,之后在40~65℃下反应3~8h,得到式(3)所示化合物;
(4)向步骤(3)所得反应体系中加酸酸化至ph值为2~6,析出固体,过滤,得到固体为所述式(4)所示的羟肟酸类捕收剂。
优选的,所述催化剂dbu与所述式(1)所示的反应物酯的质量比为1:(50~1000)。
优选的,所述式(2)所示的羟胺盐、碱moh与所述式(1)所示的反应物酯的摩尔比为:反应物酯:羟胺盐:碱moh=1:(0.5~1.5):(1.5~3),进一步优选为,反应物酯:羟胺盐:碱moh=1:(0.75~1.5):(2~3)。
作为一种优选的实施方案,步骤(3)中,所述碱溶液分两批加入到所述混合液a中,第一批碱溶液加入时,控制反应体系温度低于30℃,第一批碱溶液加完后开始体系升温,升温同时加入第二批碱溶液,在第二批碱溶液加完后控制反应温度为40~65℃,反应时间3~8h,优选3~5h。第一批碱溶液用来中和羟胺盐里的无机酸,将羟胺变成单体游离出来,中和反应剧烈,宜缓慢加入;第二批碱用来为反应提供强碱环境,可以快速加入,使反应体系迅速达到肟化反应所需的ph值,可以缩短反应时间,减少酯在碱性条件下的水解。
优选地,步骤(4)中,酸化采用浓硫酸。
优选地,所述r1选自苯基、环己基、取代的苯基、取代的环己基,取代基为卤素、c1-6的烷基、c3-6的环烷基中的任意一种。
进一步优选地,所述r1选自苯基、环己基、取代的苯基,取代基为c1-6的烷基或c3-6的环烷基,取代基更优选为戊基。
现有的羟胺法制备羟肟酸的核心过程为酯与羟胺的反应,是典型的非均相反应,酯为油相,羟胺在水相中,造成反应转化率低等问题。
本发明提供了一种全新的羟肟酸类捕收剂的合成方法,采用双环相转移催化剂dbu,其结构如下:
通过将反应物酯与双环状相转移催化剂dbu预先混合,dbu特有的分子结构使其能够充分起到相转移催化的作用,尤其是针对本发明的含有环状基团的反应物酯,可以提高油相中的酯与水相中羟胺的分子碰撞几率,从而加快反应速率、提升反应的转化率。本发明的方法具有反应速度快,转化率高的优点。
目前在羟肟酸捕收剂的制备技术上还未见有涉及双环化合物的催化合成报道。本发明提出了将含环状的原料酯与双环状相转移催化剂1,8-二氮杂二环十一碳-7-烯(dbu)混合来制备羟肟酸类捕收剂,并通过对反应步骤、反应条件等的研究,得到了一种合成羟肟酸类捕收剂的新方法,本方法反应速度快,转化率高,此外还具有反应条件温和、环保安全、后处理简单、废物产生少的优点。
附图说明
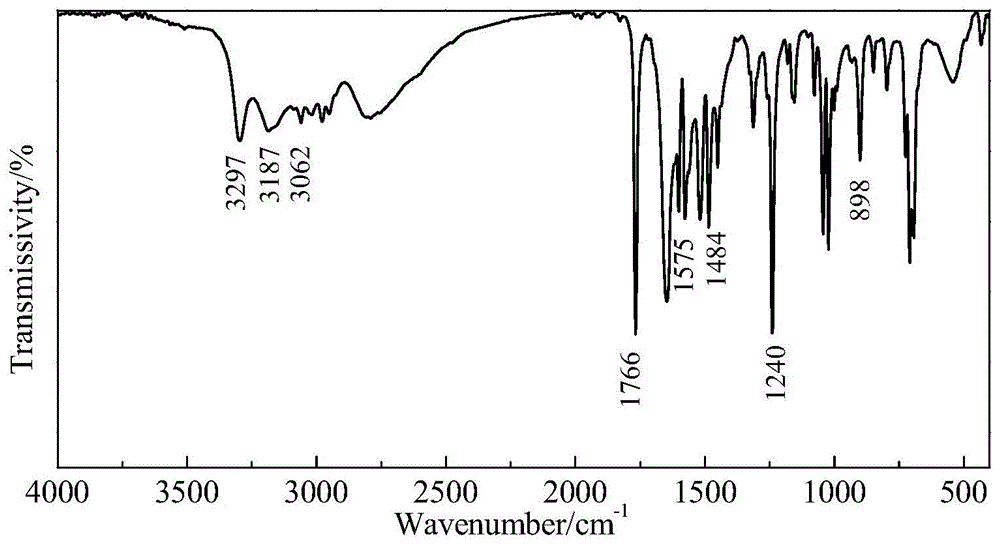
图1是苯甲羟肟酸的红外光谱;
图2是对戊基苯甲羟肟酸的红外光谱;
图3是环己甲基羟肟酸的红外光谱。
具体实施方式
下面通过实施例,对本发明的技术方案进行详细说明。
实施例1
目标化合物苯甲羟肟酸,结构式为:
反应方程式为:
通过以下方法合成:
称取0.1mol苯甲酸甲酯(13.6g)和0.014gdbu,混合搅拌5min;称取0.115mol盐酸羟胺,配制成质量分数为50%的水溶液,倒入反应容器中,之后向该反应容器加入苯甲酸甲酯和dbu的混合物,置于恒温水浴;然后称取0.22mol氢氧化钠,加水配制成质量分数为40%的碱溶液,分两批加入上述反应容器中,第一批11.5g碱溶液缓慢滴加,加完后开始升温至40℃,加入剩余的碱溶液,然后搅拌反应3小时,自然冷却至室温;用浓硫酸将产物酸化至ph为2,过滤,产品为淡红色粉末。计算转化率为84.6%。所得产物的红外光谱见图1。
实施例2
目标化合物对戊基苯甲羟肟胺,结构式为:
反应方程式为:
通过以下方法合成:
称取0.1mol对戊基苯甲酸甲酯(20.6g)和0.412gdbu,混合搅拌1h;称取0.075mol硫酸羟胺,配制成质量分数为20%的水溶液,倒入反应容器中,之后向该反应容器加入对戊基苯甲酸甲酯和dbu的混合物,置于恒温水浴;然后称取0.3mol氢氧化钾,加水配制成质量分数为20%的碱溶液,分两批加入上述反应容器,第一批42g碱溶液缓慢滴加,加完后开始升温至65℃,同时加入剩余的碱溶液,搅拌反应5小时,然后自然冷却至室温;用浓硫酸将前步产物酸化至ph为6,过滤,产品呈白色粉末状,计算转化率为77.2%。所得产物的红外光谱见图2。
实施例3
目标化合物环己甲基羟肟酸,结构式为:
反应方程式为:
通过以下方法合成:
称取0.1mol环己甲酸甲酯(14.2g)和0.0356gdbu,混合搅拌45min;称取0.125mol盐酸羟胺,配制成质量分数为40%的水溶液,倒入反应容器,之后向该反应容器加入环己甲酸甲酯和dbu的混合物;然后称取0.235mol氢氧化钠,加水配制成质量分数为40%的碱溶液,分两批加入反应容器,第一批12.5g碱溶液缓慢滴加,加完后开始升温至50℃,同时加入剩余的碱溶液,搅拌反应5小时,然后自然冷却至室温;用浓硫酸将前步产物酸化至ph为5,过滤后制得白色粉末状捕收剂产品,计算转化率为83.5%。所得产物的红外光谱见图3。
对比例1
该对比例为不使用dbu的情况下合成目标化合物环己甲基羟肟酸,通过以下方法合成:
分别称取0.1mol环己甲酸甲酯、0.125mol盐酸羟胺和0.235mol氢氧化钠,盐酸羟胺配制成质量分数为40%的水溶液,与环己甲酸甲酯一起倒入反应容器;氢氧化钠配制成质量分数为40%的水溶液,逐渐滴加至反应容器,加完后升温至50℃,搅拌反应6小时,然后自然冷却至室温;用浓硫酸将前步产物酸化至ph为5,过滤,产品为淡黄色粉末,计算转化率为56.7%。
对比例2
该对比例为不使用dbu的情况下合成目标化合物环己甲基羟肟酸,通过以下方法合成:
分别称取0.1mol环己甲酸甲酯、0.125mol盐酸羟胺和0.235mol氢氧化钠,盐酸羟胺配制成质量分数为40%的水溶液,与环己甲酸甲酯一起倒入反应容器;氢氧化钠配制成质量分数为40%的水溶液,逐渐滴加至反应容器,加完后升温至50℃,搅拌反应12小时,然后自然冷却至室温;用浓硫酸将前步产物酸化至ph为5,过滤,产品为淡黄色粉末,计算转化率为58.2%。
对比例3
目标化合物环己甲基羟肟酸,通过以下方法合成:
分别称取0.1mol环己甲酸甲酯、0.125mol盐酸羟胺、0.235mol氢氧化钠和0.0356gdbu;
盐酸羟胺配制成质量分数为40%的水溶液,与环己甲酸甲酯、dbu一起倒入反应容器;氢氧化钠配制成质量分数为40%的水溶液,逐渐滴加至反应容器,加完后升温至50℃,搅拌反应6小时,然后自然冷却至室温;用浓硫酸将前步产物酸化至ph为5,过滤,产品为淡黄色粉末,计算转化率为78.4%。
通过实施例3和对比例1-对比例3的比较,可以看出,dbu的引入能够明显提高羟肟酸合成反应的转化率,且酯与dbu通过预先混合均匀后,更有利于提高整个反应过程的均相程度,进一步促进酯向羟肟酸的转化,提高反应转化率。
以上所述仅为本发明的实施例,并非因此限制本发明的专利范围,凡是利用本发明说明书内容所作的等效变换,或直接或间接运用在其他相关的技术领域,均包括在本发明的专利保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!