左旋西替利嗪的制备方法与流程
本发明属于药物化学技术领域,尤其涉及一种左旋西替利嗪的制备方法。
背景技术:
盐酸左西替利嗪,又称左旋西替利嗪盐酸盐,化学名称为:(-)-[2-[4-(r)-4-氯苯基-苯甲基-1-哌嗪基]乙氧基]乙酸二盐酸盐;cas登记号为130018-87-0;英文通用名为levocetirizinedihydrochloride。其结构式如下:
左旋西替利嗪盐酸盐是比利时ucb公司开发的第三代h1受体拮抗剂,2001年2月在德国首次上市,商品名xyzal,左旋西替利嗪盐酸盐是盐酸西替利嗪的r型光学异构体,在临床上克服了混旋盐酸西替利嗪中由s型光学异构体引起的嗜睡、镇静等中枢神经系统副作用,用于治疗呼吸系统、眼和皮肤的过敏性疾病,适用人群广泛,且可用于孕妇与儿童,在临床应用上很受欢迎。
现有技术公开了多种左旋西替利嗪的合成方法,如cossement等(processforpreparationofa1-piperazine-ethoxyaceticacid.gb2225321,1990-05-30)报道了用化学拆分的方法拆分混旋4-氯二苯甲基哌嗪的方法。混旋4-氯二苯甲基哌嗪经l-(+)酒石酸拆分得左旋4-氯二苯基甲基哌嗪,拆分收率仅为12.7%,再与2-氯乙氧基乙腈进行烷基化反应,然后经水解、成盐等得到目标化合物左旋西替利嗪二盐酸盐。以左旋-4-氯二苯甲基哌嗪为起始原料计,总收率57.3%,光学纯度95%。但该合成路线较长。合成路线如下所示:
技术实现要素:
有鉴于此,本发明要解决的技术问题在于提供一种左旋西替利嗪的制备方法,本发明提供的制备方法路线较短,收率和纯度较高。
本发明提供了一种左旋西替利嗪的制备方法,包括以下步骤:
步骤1、式(i)所示化合物在还原体系和l-酒石酸的作用下进行反应,得到式(ii)所示化合物;
步骤2、式(ii)所示化合物在nah、n’n-二甲基甲酰胺(dmf)和四丁基溴化铵的作用下与式(iii)化合物反应,得到左旋西替利嗪。
本发明以式(i)化合物和式(iii)化合物为原料制备左旋西替利嗪,首先将式(i)化合物脱保护后进行消旋,获得式(ii)化合物;式(ii)化合物与式(iii)化合物反应即可得到左旋西替利嗪。本发明提供的方法反应路线较短,收率可达54%以上,纯度最高可达99.65%。
在本发明中,式(i)所示化合物按照以下方法制备:
式(iv)化合物、对氯苯甲醛和溴苯在催化剂作用下反应,得到式(i)化合物;
其中,式(iv)化合物按照以下方法制备:
哌嗪和对甲苯磺酰氯在三乙胺和二氯甲烷(dcm)中反应,得到式(iv)化合物。
具体而言,将哌嗪、对甲苯磺酰氯、三乙胺和二氯甲烷混合后进行反应,得到式(iv)化合物。其中,所述反应的温度为20~25℃,时间为2~4h。所述哌嗪、对甲苯磺酰氯、三乙胺和二氯甲烷的质量比为40~60:120~160:60~100:350~450。
得到式(iv)化合物后,将其与对氯苯甲醛、溴苯和催化剂混合反应,得到式(i)化合物。具体而言,所述反应的温度为70~75℃,时间为4~6h。所述反应在乙腈中进行。所述催化剂选自溴化钴、溴化锌或锌粉。式(iv)化合物、对氯苯甲醛、溴苯和催化剂的质量比为45~55:25~35:30~40:1~3。
式(i)化合物在还原体系和l-酒石酸的作用下进行反应,得到式(ii)所示化合物。
具体而言,首先将式(i)化合物在还原体系中进行脱保护,然后将其在l-酒石酸的作用下进行消旋。
在一个实施例中,所述还原体系选自溴化氢和乙酸。将式(i)化合物、溴化氢和乙酸混合进行脱保护,所述反应的温度为10~15℃,时间为2~4h。
脱保护后向得到的产品中加入甲醇和l-酒石酸,消旋后得到式(ii)化合物。所述反应的温度为60~65℃,反应时间为1~2h。
在本发明中,式(iii)化合物按照以下方法制备:
1,4-二氧六环在单过硫酸氢钾复合盐的作用下进行反应,得到式(iii)化合物。
1,4-二氧六环和单过硫酸氢钾复合盐在二氯亚砜中进行反应,得到式(iii)化合物。其中,所述反应温度为20~25℃,时间为3~5h。
得到(iii)化合物后,在nah、dmf和四丁基溴化铵的作用下与式(iii)化合物反应,即可得到左旋西替利嗪。
具体而言,将式(ii)化合物、式(iii)化合物、四丁基溴化铵和dmf加入反应瓶中,氮气保护下加入氢化钠,反应即可。其中,所述反应的温度为90~95℃,时间为4~6h。
本发明以式(i)化合物和式(iii)化合物为原料制备左旋西替利嗪,首先将式(i)化合物脱保护后进行消旋,获得式(ii)化合物;式(ii)化合物与式(iii)化合物反应即可得到左旋西替利嗪。本发明提供的方法反应路线较短,收率可达54%以上,最高可达86.5%,纯度可达95.5%以上,最高可达99.65%。
附图说明
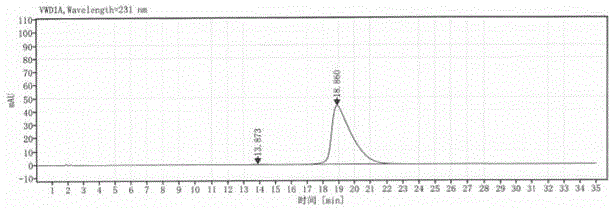
图1是本发明实施例提供的左旋西替利嗪的高效液相色谱图。
具体实施方式
下面将结合本发明实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
以下实施例中所用的试剂均为市售。
实施例1
步骤1、将60g1,4-二氧六环、89.1g二氯亚砜和30g单过硫酸氢钾复合盐加入反应瓶中,反应温度为25℃,时间为4h,gc检测二氧六环反应完全,蒸馏后得到产物式(iii)化合物,经gc检测纯度为98.6%,收率96.1%。
步骤2、将60g哌嗪、146.1g对甲苯磺酰氯、84.6g三乙胺和397.5g二氯甲烷加入反应瓶中,反应温度为25℃,时间为3h,tlc检测哌嗪反应完全,加水淬灭,有机相用60g水洗涤两次,45℃浓缩至无液滴滴出,得到式(iv)化合物,纯度95.4%,收率91.2%。
步骤3、将50g化合物式(iv)、30.7g对氯苯甲醛、34.3g溴苯和300ml乙腈加入反应瓶中,氮气保护,升温至75℃,在2.5g催化剂溴化钴作用下反应5h。tlc检测原料反应完全,过滤,滤液浓缩至小体积,加入100ml甲醇,浓缩至小体积粘稠状,再加入50ml甲醇,室温搅拌0.5h,过滤,得到化合物式(i),收率86.5%,纯度91.5%。
步骤4、将50g化合物式(i)加入250g二氯甲烷中,氮气保护,降温至10℃,再加入11g溴化氢、8.2g乙酸,反应3h,tlc检测原料反应完全,加入200g8%碳酸氢钠溶液,萃取分液,有机相浓缩至小体积,加入500ml甲醇,l-酒石酸16.5g,升温至65℃,反应2h,降温析晶,过滤,固体加水溶解,用碱调ph至11,加200ml二氯甲烷萃取两次,合并有机相,加水100ml洗涤,有机相浓缩至干,加150ml正己烷搅拌析晶,过滤,干燥,得到化合物式(ii),收率82.3%,纯度93.6%。
步骤5、将20g化合物式(ii)、10.6g化合物式(iii)、1.1g四丁基溴化铵、80mldmf加入反应瓶中,氮气保护下加入4.1g氢化钠,升温至95℃,反应时间为5h,tlc检测原料反应完全,将反应液倒入600ml水中,室温搅拌1h,过滤,得到左旋西替利嗪,收率86.5%,纯度95.5%。
参见图1和表1,图1是本发明实施例提供的左旋西替利嗪的高效液相色谱图,表1为本发明实施例提供的左旋西替利嗪的高效液相色谱数据。
表1本发明实施例提供的左旋西替利嗪的高效液相色谱数据
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!