新型单硫醇粘液溶解剂的制作方法
新型单硫醇粘液溶解剂
[0001]
本申请是中国专利申请号201680013820.1(pct/us2016/015353),申请日2016年1月28日,发明名称为“新型单硫醇粘液溶解剂”的分案申请。
[0002]
继续申请信息
[0003]
本申请要求于2015年1月30日提交的美国临时申请系列no.62/109,999的优先权,并且通过引入并入本文。
[0004]
发明背景
发明领域
[0005]
本发明涉及新型单硫醇粘液溶解剂。本发明还包括使用这些创新的粘液溶解剂的各种治疗方法。
[0006]
背景说明
[0007]
在环境与身体之间的界面处的粘膜表面已经形成了许多“天生防御”,即保护机制。粘液运输系统是呼吸道对于吸入的颗粒/感染因子的基本防御。吸入的颗粒被捕获在粘液层中,随后通过粘液清除从肺中推出。粘液运输系统要求粘液充分水合以促进纤毛清除(cilliary clearance)。在没有足够的粘液水合作用的情况下,粘液变得过度粘稠和黏附,这可能导致气道粘液积聚和感染。
[0008]
通常,粘膜表面上的液体层的数量反映了上皮液体分泌物与上皮液体吸收之间的平衡,上皮液体分泌物通常反映与水(和阳离子抗衡离子)偶联的阴离子(cl-和/或hco
3-)分泌,上皮液体吸收通常常反映与水和抗衡阴离子(cl-和/或hco
3-)偶联的na
+
吸收。粘膜表面的许多疾病是由分泌物(太少)和吸收(相对太多)之间的不平衡造成的那些粘膜表面上太少的保护性液体引起的。表征这些粘膜功能障碍的缺陷盐转运过程存在于粘膜表面的上皮层中。
[0009]
粘液运输系统异常表征包括囊性纤维化(cf)和慢性支气管炎(cb)在内的粘膜阻塞性气道疾病的复杂性。正常的粘液清除需要1)气道表面的充分水合和2)粘液与细胞表面之间没有强烈的粘合和凝聚的相互作用。
[0010]
水合作用由纤周和粘液层中的粘蛋白浓度定义。离子传输性质调节盐和水(即溶剂)的量,杯状细胞和腺体控制气道表面上的粘蛋白的量。具有粘液阻塞性疾病(包括囊性纤维化(cf))、与香烟烟雾暴露相关的慢性支气管炎(即copd)和哮喘的受试者表现出由固体百分比含量定量的粘液浓度增加(参见图1),这是由于气道水合减少和粘蛋白分泌过多,导致杯状细胞和腺体增生引起的。两者作为疾病严重程度的函数,在急性加重中,升高的粘蛋白浓度会产生粘附于上皮细胞的粘附粘液,损害清除,引发炎症反应和气道壁损伤,并且用作致病微生物的生长培养基。显然,增加这些增厚/粘附的粘液从气道的清除可能有益于患有粘液性阻塞性疾病的患者。
[0011]
慢性支气管炎(cb),包括最常见的致死性遗传形式的慢性支气管炎,囊性纤维化(cf),是反映身体未能清除通常来自肺部粘液,最终导致慢性气道感染的疾病。在正常肺中,慢性肺内呼吸道感染(慢性支气管炎)的主要防御措施是由支气管气道表面的粘液的连
续清除所介导。健康的这个功能有效地从肺部去除潜在的有毒毒素和病原体。最近的数据表明,cb和cf两者的起始问题,即“基本缺陷”是未能从气道表面清除粘液。清除粘液的失败反映了气道表面上液体与粘蛋白的量之间的不平衡。这种“气道表面液体”(asl)主要由盐和水以与血浆相类似的比例(即等渗)组成。粘蛋白大分子组织成一个明确定义的“粘液层”,其通常会捕获吸入的细菌,并通过在称为“纤周液体(periciliary liquid)”(pcl)的水溶性、低粘度溶液中打碎的纤毛的作用将其输送出肺。在疾病状态下,气道表面上的粘液与asl的量存在不平衡。这导致asl的相对减少,其导致粘液浓缩,pcl的润滑剂活性的降低,以及未能通过对嘴的睫状肌活性将粘液清除至嘴。来自肺的粘液的机械清除率的降低导致粘附于气道表面的粘液的慢性细菌定居。这是细菌的长期保留,局部抗菌物质以慢性方式杀死粘液捕获的细菌的失效,以及随之而来的身体对这种类型的表面感染的慢性炎症反应,这导致cb和cf的综合征。
[0012]
目前在美国的受累人群是1200万具有慢性支气管炎的获得性(主要来自香烟烟雾暴露)形式的患者,和约30,000名具有遗传形式,囊性纤维化的患者。在欧洲存在大致相同的这两种人群的数量。在亚洲,只有少量cf,但cb的发病率很高,像世界其它地区一样,发病率也在上升。
[0013]
目前,存在对在导致这些疾病的基本缺陷的水平上专门治疗cb和cf的产品的大量未满足的医疗需求。目前用于慢性支气管炎和囊性纤维化的治疗集中在治疗这些疾病的症状和/或晚期作用。因此,对于慢性支气管炎,β-激动剂、吸入类固醇、抗胆碱能药以及口服茶碱和磷酸二酯酶抑制剂都在开发中。然而,这些药物中没有一种有效地治疗未能从肺部清除粘液的根本问题。类似地,在囊性纤维化中,使用相同谱的药理剂。这些策略已已得到了更新策略的补充,所述策略被设计来通过中性粒细胞(所述中性粒细胞试图杀死在粘附粘液块中生长的细菌)以及通过使用吸入抗生素(“tobi”)(所述抗生素被设计来增加肺部自身的杀伤机制,以消除细菌的粘附粘液斑块)清除cf肺的已沉积在肺中的dna(“pulmozyme”;genentech)。身体的一般原则是,如果起始病变没有得到治疗,在这种情况下,粘液滞留/阻塞,细菌感染变成慢性且抗微生物治疗越来越难以治疗。因此,cb和cf肺部疾病的主要未满足的治疗需求是动员气道粘液并促进其与细菌从肺部清除的有效手段。
[0014]
身体内和身体上的其它粘膜表面表现出保护性表面液体在其表面上的正常生理学上的微妙差异,但疾病的病理生理学反映出共同的主题,即保护性表面液体太少和粘液清除受损。例如,在口腔干燥(口干)中,口腔由于腮腺舌下和下颌下腺分泌液体的失败而耗尽液体。类似地,干燥性角膜结膜炎(干眼症)由因泪腺不能分泌液体或过度的蒸发液体损失而导致的泪液体积不足引起。在鼻鼻窦炎中,如在cb中,在粘蛋白分泌、相对气道表面液体消耗和粘液淤滞之间存在不平衡。最后,在胃肠道中,不能在近端小肠中分泌c1-(和液体)结合末端回肠的增加的na
+
(和液体)吸收导致远端肠梗阻综合征(dios)。在老年患者中,下降结肠中过量的na
+
(和体积)吸收导致便秘和憩室炎。
[0015]
急性支气管炎和慢性支气管炎的高发病率表明,这种疾病综合征是美国的主要健康问题。尽管粘液阻塞性疾病的病因学取得显著进步,但cf和copd的药物治疗的特征在于老化的治疗方法,通常包括用于维护的吸入类固醇和支气管扩张剂,以及用于恶化的抗生素和高剂量类固醇。显然,需要的是更有效地恢复从cb/cf患者肺部清除粘液的药物。这些新疗法的价值将反映在cf和cb人群的生活质量和持续时间的改善。
[0016]
增加粘液清除的一种方法是通过破坏聚合物粘液结构来提高粘蛋白的输送性。粘蛋白蛋白通过共价(二硫化物)和非共价键的形成被组织成高分子量聚合物。利用还原剂破坏共价键是在体外降低粘液的粘弹性的已知方法,并且经预测最小化粘液粘附性并在体内改善清除率。众所周知,还原剂可在体外降低粘液的粘度,并通常用作处理痰样品的辅助手段(hirsch,s.r.,zastrow,j.e.和kory,r.c.sputum liquefying agents:a comparative in vitro evaluation.j.lab.clin.med.1969.74:346-353)。还原剂的实例包括含有能够还原蛋白质二硫键的分子的硫化物,包括但不限于n-乙酰半胱氨酸、n-acystelyn、羧甲司坦、半胱胺、谷胱甘肽和含硫氧还蛋白的蛋白质。
[0017][0018]
n-乙酰半胱氨酸(nac)被批准与胸部物理治疗结合使用以松动粘稠或增稠的气道粘液。评估口服或吸入nac在cf和copd中作用的临床研究报告了粘液的流变学特性的改善以及朝向肺功能改善的趋势和肺部恶化的减少(duijvestijn ycm和brand plp.;systematic review of n-acetylcysteine in cystic fibrosis.acta peadiatr 88:38-41.1999)。然而,临床数据的优势表明,当口服给药或作为吸入气雾剂时,nac最多是治疗气道粘液阻塞的边缘有效治疗剂。最近cochrane对现有的关于使用nac的临床文献的综述发现没有证据支持nac对cf的疗效(nash ef,stephenson a,ratjen f,tullis e.;nebulized and oral thiol derivatives for pulmonary disease in cystic fibrosis.cochrane database syst rev.2009;21(1):cd007168.)。
[0019]
作为局部肺部治疗剂的nac对于还原粘蛋白二硫键不是最佳的。具体地说,nac不具有一个有效肺部药物的基本性质,因为nac(1)在气道表面环境(例如,cf ph 6.5-7.2)中是相对低效的还原剂;和(2)被快速代谢并从气道表面清除(jayaraman s,song y,vetrivel l,shankar l,verkman as.noninvasive in vivo fluorescence measurement of airway-surface liquid depth,salt concentration,and ph.j clin invest.2001;107(3):317-24)。例如,在气道表面的ph环境(在cf和copd气道中经测量在ph 6.0至7.2的范围内),nac仅部分以其反应状态作为带负电荷硫醇盐的存在(jayaraman s,song y,vetrivel l,shankar l,verkman as.noninvasive in vivo fluorescence measurement of airway-surface liquid depth,salt concentration,and ph.j clin invest.2001;107(3):317-24)(图3)。此外,在动物研究中,通过吸入给药的
14
c-标记的nac显示出以约20分钟的半衰期从肺部快速清除(未发表的观察)。nac在生理气道ph值下的相对低的还原活性和nac在肺表面上的短的半衰期为在粘液阻塞性疾病中缺乏有效粘液还原的强有力临床证据提供了解释。
[0020]
另外,最常见地将nac作浓缩吸入溶液(是20%或1.27m的溶液)进行施用。然而,浓缩的nac溶液的施用会影响nac的耐受性,因为其增强了(1)令人不快的硫味/
气味;和(2)肺部副作用,包括刺激性和支气管收缩,这可能需要联合施用抢救药物诸如支气管扩张剂。虽然痰易净(mucomyst)在1963年被fda批准,但目前没有其它作为吸入气雾剂的还原剂可用于治疗粘膜阻塞性疾病。需要的是用于治疗以粘液清除受损为特征的疾病的有效、安全和被良好耐受的还原剂。
技术实现要素:
[0021]
本发明的一个目的涉及一种增加患者的粘液液化的方法,所述的患者具有过多粘液或具有增加的粘弹性、粘性或粘附性性质的粘液。该方法包括使具有异常或过多粘液的患者的粘液与包含含有二硫醇基团的粘液溶解化合物的组合物接触,以通过还原粘蛋白二硫键来降低粘液粘弹性的步骤。
[0022]
本发明的目的是提供粘液溶解化合物,其比n-乙酰半胱氨酸(nac)更有效和/或从粘膜表面更少地快速被吸收和/或耐受性更好。
[0023]
本发明的另一个目的是提供在气道表面的生理环境中更具活性的化合物。
[0024]
本发明的另一个目的是提供化合物。该化合物与诸如n-乙酰半胱氨酸的化合物相比,更强效和/或被更少地快速被吸收。
[0025]
因此,与nac相比,这样的化合物将在粘膜表面上产生延长的药效半衰期。
[0026]
本发明的另一个目的是提供利用上述化合物的药理学特性的治疗方法。
[0027]
特别地,本发明的目的是提供依赖于促进从粘膜表面清除粘液的治疗方法。
[0028]
本发明的目的是提供比已知化合物更强效和/或从粘膜表面更少地快速被吸收和/或更少可逆的化合物。
[0029]
因此,与已知化合物相比,所述化合物将在粘膜表面上产生延长的药效半衰期。
[0030]
本发明的另一个目的是提供与已知化合物相比,(1)从粘膜表面,特别是气道表面更少地快速被吸收的化合物;(2)本发明的另一个目的是提供与化合物(诸如nac)相比,更强效和/或被更少地快速被吸收和/或表现出更少的可逆性化合物。因此,与先前的化合物相比,这样的化合物将在粘膜表面上产生延长的药效半衰期。
[0031]
本发明的另一个目的是提供利用上述化合物的药理学特性的治疗方法。
[0032]
特别地,本发明的目的是提供依赖于粘膜表面再水合的治疗方法。
[0033]
本发明的目的可用包含结构(ia)-(id)的式i化合物表示的一类二硫醇来实现:
cap、-连接基-(ch2)
n-(z)
g-(ch2)
m-(z)
g-cap或-连接基-z
g-(ch2)
m-het-(ch2)
m-cap,
[0040]
条件是至少一个r5基团含有至少一个碱性氮;
[0041]
每个r6独立地是氢、-c(=o)-r7或天然氨基酸构型的氨基酰基;
[0042]
每个r7独立地是氢,低级烷基,苯基,取代的苯基,低级烷基苯基、-ch2(chor8)
m-ch2or8、2-呋喃基或3-呋喃基;
[0043]
每个r8独立地是氢、低级烷基、低级烷基苯基、-c(=o)-r
11
、葡糖苷酸、2-四氢萘基或
[0044][0045]
每个r9独立地是-co2r7、-con(r7)2、-so2ch3、-c(=o)r7、-co2r
13
、-con(r
13
)2、-so2ch2r
13
或-c(=o)r
13
;
[0046]
每个r
10
独立地是-h、-so2ch3、-co2r7、-c(=o)nr7r9、
[0047]-c(=o)r7或-ch
2-(choh)
n-ch2oh;
[0048]
每个z独立地是-(choh)-、-c(=o)-、-(chnr7r
10
)-、-(c=nr
10
)-、-nr
10-、-(ch2)
n-,-(chnr
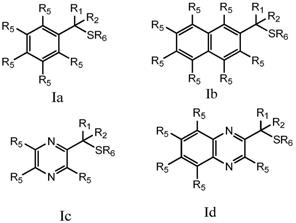
13
r
13
)-、-(c=nr
13
)-或-nr
13
、-co2h;
[0049]
每个r
11
独立地是氢、低级烷基、苯基低基烷基或取代的苯基低级烷基;
[0050]
每个r
12
独立地是-so2ch3、-co2r7、-c(=o)-nr7r9、-c(=o)-r7、-ch2(choh)
n-ch2oh、-co2r
13
、-c(=o)-nr
13
r
13
或-c(=o)r
13
;
[0051]
每个r
13
独立地是氢、低级烷基、苯基、取代的苯基或-ch2(chor8)
m-ch2or8、-so2ch3、-co2r7、-c(=o)nr7r9、-c(=o)-r7、-ch
2-(choh)
n-ch2oh、-(ch2)
m-nr7r
10
、-(ch2)
m-nr7r7、-(ch2)
m-nr
11
r
11
、-(ch2)
m-(nr
11
r
11
r
11
)
+
、-(ch2)
m-(chor8)
m-(ch2)
m
nr
11
r
11
、-(ch2)
m-(chor8)
m-(ch2)
m
nr7r
10
、-(ch2)
m-nr
10
r
10
、-(ch2)
m-(chor8)
m-(ch2)
m-(nr
11
r
11
r
11
)
+
、-(ch2)
m-(chor8)
m-(ch2)
m
nr7r7;
[0052]
每个g独立地是1至6的整数;
[0053]
每个m独立地是1至7的整数;
[0054]
每个n独立地是0至7的整数;
[0055]
每个-het-独立地是-n(r7)-,-n(r
10
)-、-s-、-so-、-so
2-;-o-、-so2nh-、-nhso
2-、-nr7co-、-conr
7-、-n(r
13
)-、-so2nr
13-、-nr
13
co-或-conr
13-;
[0056]
每个连接基独立地是-o-、-(ch2)
n-、-o(ch2)
m-、-nr
13-c(=o)-nr
13-、-nr
13-c(=o)-(ch2)
m-、-c(=o)nr
13-(ch2)
m-、-(ch2)
n-(z)
g-(ch2)
n-、-s-、-so-、-so
2-、-so2nr
7-、-so2nr
10-或-het-;
[0057]
每个cap独立地是:
[0058]
[0059][0060]
条件是当任何-chor
8-或-ch2or8基团相对于彼此位于1,2或1,3-时,r8基团可以任选地一起形成环状单取代或二取代的1,3-二噁烷或1,3-二氧戊环;和外消旋物、对映异构
体、非对映异构体、互变异构体、多晶型物、假多晶型物及其药学上可接受的盐。
[0061]
本发明还提供包含本文所述的化合物的药物组合物。
[0062]
本发明还提供恢复粘膜防御的方法,其包括:
[0063]
对有此需要的受试者使粘液与有效量的本文所述化合物相接触。
[0064]
本发明还提供了降低粘液粘弹性的方法,其包括:向受试者的粘膜表面施用效量的本文所述的化合物。
[0065]
本发明还提供了在粘膜表面上降低粘液粘弹性的方法,其包括:向受试者的粘膜表面施用有效量的本文所述的化合物。
[0066]
本发明还提供清除粘膜表面上的自由基的方法,其包括:向受试者的粘膜表面施用有效量的本文所述的化合物。
[0067]
本发明还提供减少粘膜表面上的炎症的方法,其包括:向受试者的粘膜表面施用有效量的本文所述的化合物。
[0068]
本发明还提供了在粘膜表面上淬灭氧化自由基的方法,其包括:向受试者的粘膜表面施用有效量的本文所述的化合物。
[0069]
本发明还提供了减少粘膜表面上的炎症细胞的方法,其包括:向受试者的粘膜表面施用有效量的本文所述的化合物。
[0070]
本发明还提供了治疗粘液阻塞性疾病的方法,其包括:对有此需要的受试者使粘液与有效量的本文所述化合物相接触。
[0071]
本发明还提供了治疗粘液粘附的方法,其包括:对有此需要的受试者使粘液与有效量的本文所述化合物相接触。
[0072]
本发明还提供了治疗肺纤维化的方法,其包括:向有此需要的受试者施用有效量的本文所述的化合物。
[0073]
本发明还提供了治疗慢性支气管炎的方法,其包括:向有此需要的受试者施用有效量的本文所述的化合物。
[0074]
本发明还提供一种治疗囊性纤维化的方法,其包括:向有此需要的受试者施用有效量的本文所述的化合物。
[0075]
本发明还提供了治疗囊性纤维化恶化的方法,其包括:向有此需要的受试者施用有效量的本文所述的化合物。
[0076]
本发明还提供了治疗肺纤维化的方法,其包括:向有此需要的受试者施用有效量的本文所述的化合物。
[0077]
本发明还提供了治疗支气管扩张症的方法,其包括:向有此需要的受试者施用有效量的本文所述的化合物。
[0078]
本发明还提供了治疗慢性阻塞性肺疾病的方法,其包括:向有此需要的受试者施用有效量的本文所述的化合物。
[0079]
本发明还提供了治疗慢性阻塞性肺疾病恶化的方法,其包括:向有此需要的受试者施用有效量的本文所述的化合物。
[0080]
本发明还提供了治疗哮喘的方法,其包括:向有此需要的受试者施用有效量的本文所述的化合物。
[0081]
本发明还提供了治疗哮喘恶化的方法,其包括:向有此需要的受试者施用有效量
的本文所述的化合物。
[0082]
本发明还提供了治疗食管炎的方法,其包括:向有此需要的受试者施用有效量的本文所述的化合物。
[0083]
本发明还提供了治疗呼吸机诱发性肺炎的方法,包括:通过呼吸机向受试者施用本文所述的有效化合物。
[0084]
本发明还提供了治疗原发性纤毛运动障碍的方法,其包括:向有此需要的受试者施用有效量的本文所述的化合物。
[0085]
本发明还提供了治疗肺气肿的方法,其包括:向有此需要的受试者施用有效量的本文所述的化合物。
[0086]
本发明还提供了治疗肺炎的方法,其包括:向有此需要的受试者施用有效量的本文所述的化合物。
[0087]
本发明还提供了一种治疗鼻鼻窦炎的方法,包括:向有需要的受试者施用有效量的本文所述的化合物。
[0088]
本发明还提供了治疗鼻脱水的方法,其包括:向有此需要的受试者的鼻道施用有效量的本文所述的化合物。
[0089]
在具体实施方案中,通过向受试者施用干氧来产生鼻脱水。
[0090]
本发明还提供了治疗鼻窦炎的方法,其包括:向有此需要的受试者施用有效量的本文所述的化合物。
[0091]
本发明还提供了治疗便秘的方法,其包括:向有此需要的受试者施用有效量的本文所述的化合物。在该方法的一个实施方案中,口服或通过栓剂或灌肠剂施用所述化合物。
[0092]
本发明还提供了治疗远端肠梗阻综合征的方法,其包括:向有此需要的受试者施用有效量的本文所述的化合物。
[0093]
本发明还提供了治疗慢性憩室炎的方法,其包括:向有此需要的受试者施用有效量的本文所述的化合物。
[0094]
本发明还提供了为了诊断目的诱导痰的方法,其包括:向有此需要的受试者施用有效量的本文所述的化合物。
[0095]
本发明还提供了治疗吸入的病原体的方法,其包括:向有此需要的受试者施用有效量的本文所述的化合物。
[0096]
本发明还提供了治疗吸入的刺激物的方法,其包括:向有此需要的受试者施用有效量的本文所述的化合物。
[0097]
本发明还提供了治疗吸入的颗粒的方法,其包括:向有此需要的受试者施用有效量的本文所述的化合物。
[0098]
在具体实施方案中,所述吸入颗粒是包括灰尘、碎屑或放射性物质的不溶性颗粒。
[0099]
本发明的目的也可以用治疗炭疽的方法来实现,包括向有此需要的受试者施用有效量的本文所定义的式i的化合物和渗透物。
[0100]
本发明的目的还可通过针对由病原体,特别是由可能用于生物恐怖主义的病原体引起的疾病或病症的预防性、暴露后预防性、预防性或治疗性治疗方法来实现,包括向有此需要的受试者施用有效量的式i的化合物。
[0101]
本发明的另一个目的是提供治疗,其包括使用渗透物与式i的粘液溶解剂,所述粘
液溶解剂与诸如nac的化合物相比更加强效,更特异和/或更少地快速从粘膜表面吸收。
[0102]
本发明的另一方面是提供使用式i的粘液溶解剂的治疗,所述粘液溶解剂当与渗透增强剂一起施用时与诸如nac的化合物相比,更加强效和/或更少地快速被吸收和/或表现更低的可逆性。因此,与单独使用的化合物相比,当与渗透物结合使用时,此类粘液溶解剂将对粘膜表面产生增强的药效学作用。
[0103]
本发明的另一个目的是提供一起使用式i的粘液溶解剂和渗透物的治疗,所述粘液溶解剂相较于nac被更少地快速从粘膜表面,特别是气道表面吸收。本发明的另一个目的是提供含有式i的粘液溶解剂和渗透物的组合物。
[0104]
本发明的目的可以通过治疗疾病的方法来实现,其中通过增加的粘液清除和粘膜水合而改善,包括向需要增加的粘液纤毛清除和/或粘膜水合的受试者施用有效量的如本文定义的式i的化合物和渗透物。
[0105]
附图简述
[0106]
图1.粘液脱水在cf/copd病原序列中的作用。
[0107]
图2.化合物76相对于nac的增强的粘蛋白还原能力。
[0108]
图3.在没有损失还原活性的情况下对化合物76的修饰。
[0109]
图4.相对于本领域已知的化合物的还原活性。
[0110]
图5.化合物76取代物的优点。
[0111]
图6.评估细胞渗透和促炎症作用的综合测定。
[0112]
图7.化合物86的前药。
[0113]
图8.化合物85在copd痰中的代谢活化。
[0114]
图9.原发性人支气管上皮(hbe)培养物上的粘液还原。
[0115]
发明详述
[0116]
如本文所用,以下术语如所示定义。
[0117]“本发明的化合物”意指式i化合物或其盐,特别是其药学上可接受的盐。
[0118]“式i的化合物”意指具有在本文中命名为式i的结构式的化合物。式i的化合物包括溶剂合物和水合物(即,式i的化合物与溶剂的加合物)。在其中式i的化合物包括一个或多个手性中心的那些实施方案中,该短语旨在涵盖包括旋光异构体(对映异构体和非对映异构体)和几何异构体(顺式-/反式-异构体)的每种单独的立体异构体以及立体异构体的混合物。此外,式i的化合物还包括所述式的互变异构体。
[0119]
在整个说明书和实施例中,化合物使用标准iupac命名原则来命名,在可能的情况下包括使用由cambridgesoft corp./perkinelmer出售的用于命名化合物的chemdraw ultra 11.0软件程序来命名。
[0120]
在其中碳原子不具有足够数量的被描绘来产生四价的所附变量的一些化学结构表示中,需要来提供四价的剩余碳取代基应假定为氢。类似地,在其中没有指定末端基团的情况下键被画出的一些化学结构中,这种键指示甲基(me,-ch3)基团,这在本领域是常规的。
[0121]
本发明基于以下发现:相较于nac和dtt,式i的化合物在粘膜表面,特别是气道表面中更加高效和/或,被更少地快速被吸收,达到更高的浓度并且具有更长的停留时间。因此,与nac相比,式i的化合物具有更大的活性和/或对粘膜表面产生更少的细胞毒性。
[0122]
本发明基于以下发现:与化合物(诸如nac)相比,式(i)的化合物更加强效和/或,从粘膜表面、特别是气道表面更少地快速被吸收,和/或更少可逆的相互作用。
[0123]
因此,与这些化合物相比,式(i)的化合物在粘膜表面上具有更长的半衰期。
[0124]
在包含结构(ia)-(id)的由式i表示的化合物中:
[0125][0126]
其中r1和r2各自独立地为氢、低级烷基、卤素、三氟甲基、羟基-低级烷基、苯基、(苯基)-低级烷基、(卤代苯基)-低级烷基、((低级烷基)苯基)-低级烷基、((低级-烷氧基)苯基)-低级烷基、(萘基)-低级烷基或(吡啶基)-低级烷基;
[0127]
r3和r4各自独立地为氢、低级烷基、羟基-低级烷基、苯基、(苯基)-低级烷基、(卤代苯基)-低级烷基、((低级-烷基)苯基)-低级-烷基、((低级烷氧基)苯基)-低级-烷基、(萘基)-低级-烷基或(吡啶基)-低级-烷基;
[0128]
每个r5独立地为氢、卤素、三氟甲基、低级烷基、未取代或取代的苯基、低级烷基-硫基、苯基-低级烷基-硫基、低级烷基-磺酰基或苯基-低级烷基-磺酰基、oh、-(ch2)
m-or8、-o-(ch2)
m-or8、-(ch2)
n-nr7r
10
、-(ch2)
n-nr7r7,-(ch2)
n-nr7r7,-o-(ch2)
m-nr7r
10
、-o-(ch2)
m-nr7r7、-o-(ch2)
m-nr7r7、-(ch2)
n
(chor8)(chor8)
n-ch2or8、-o-(ch2)
m
(chor8)(chor8)
n-ch2or8、-(ch2ch2o)
m-r8、-o-(ch2ch2o)
m-r8、-(ch2ch2o)
m-ch2ch2nr7r
10
、-o-(ch2ch2o)
m-ch2ch2nr7r
10
、-(ch2)
n-c(=o)nr7r
10
、-o-(ch2)
m-c(=o)nr7r
10
、-(ch2)
n-(z)
g-r7、-o-(ch2)
m-(z)
g-r7、-(ch2)
n-nr
10-ch2(chor8)(chor8)
n-ch2or8、-o-(ch2)
m-nr
10-ch2(chor8)(chor8)
n-ch2or8、-(ch2)
n-co2r7、-o-(ch2)
m-co2r7、-oso3h、-o-葡糖苷酸、-o-葡萄糖、
co2r7、-oso3h、-o-葡糖苷酸、-o-葡萄糖,
[0139][0140]
在优选实施方案中,每个-(ch2)
n-(z)
g-r7落入上述结构的范围内,并且独立地是-(ch2)
n-nh-c(=nh)nh2,
[0141]
在另一个优选实施方案中,每个-o-(ch2)
m-(z)
g-r7落入上述结构的范围内,并且独立地是
[0142]-o-(ch2)
m-nh-c(=nh)-n(r7)2或
[0143]-o-(ch2)
m-chnh
2-co2nr7r
10
。
[0144]
在另一个优选的实施方案中,r5是-oh、-o-(ch2)
m
(z)
g
r
12
、-het-(ch2)
m-nh-c(=nr
13
)-nr
13
r
13
、-het-(ch2)
n-(z)
g-(ch2)
m
nh-c(=nr
13
)-nr
13
r
13
、-连接基-(ch2)
m-(z)
g-(ch2)
m-cap、连接基-(ch2)
n-cr
11
r
11-cap、-het-(ch2)
m-conr
13
r
13
、-(ch2)
n-nr
12
r
12
、-o-(ch2)
m
nr
11
r
11
、-o-(ch2)
m-n
⊕-(r
11
)3、-(ch2)
n-(z)
g-(ch2)
m-nr
10
r
10
、-het-(ch2)
m-(z)
g-nh-c(=nr
13
)-nr
13
r
13
、-o-(ch2)
m
(chor8)(chor8)
n-ch2or8、-o-(ch2)
m-c(=o)nr7r
10
、-o-(ch2)
m-(z)
g-r7或-o-(ch2)
m-nr
10-ch2(chor8)(chor8)
n-ch2or8。
[0145]
在特别优选的实施方案中,r5是-连接基-(ch2)
m-cap、-连接基-(ch2)
n
(chor8)(chor8)
n-cap、-连接基-(ch2ch2o)
m-ch
2-cap、-连接基-(ch2ch2o)
m-ch2ch
2-cap、-连接基-(ch2)
m-(z)
g-cap、-连接基-(ch2)
n
(z)
g-(ch2)
m-cap、-连接基-(ch2)
n-nr
13-ch2(chor8)(chor8)
n-cap、-连接基-(ch2)
n-(chor8)
m
ch
2-nr
13-(z)
g-cap、-连接基-(ch2)
n
nr
13-(ch2)
m
(chor8)
n
ch2nr
13-(z)
g-cap、-连接基-(ch2)
m-(z)
g-(ch2)
m-cap、-连接基-nh-c(=o)-nh-(ch2)
m-cap、-连接基-(ch2)
m-c(=o)nr
13-(ch2)
m-cap、-连接基-(ch2)
n-(z)
g-(ch2)
m-(z)
g-cap或-连接基-z
g-(ch2)
m-het-(ch2)
m-cap。
[0146]
在另一个特别优选的实施方案中,r5是-o-(ch2)
m-nr
10
r
10
、-o-(ch2)
m-nr7r
10
、-(ch2)
n-nr7r
10-(ch2)
n-nr
10
r
10
,-(ch2)
n-nr7r7、-o-(ch2)
m-nr7r
10
。
[0147]
每个r6独立地是氢、-c(=o)-r7或或天然氨基酸构型的氨基酰基;
[0148]
在优选的实施方案中,r6是h、异丁酰基、丙酰基或2-糠酰基。
[0149]
在特别优选的实施方案中,r6是乙酰基。
[0150]
在另一个优选的实施方案中,r6是-(c=o)-chnh
2-(ch2)4nh2。
[0151]
天然氨基酸构型的氨基酰基是指由甘氨酸、丙氨酸、缬氨酸、亮氨酸、异亮氨酸、半胱氨酸、甲硫氨酸、苯丙氨酸、酪氨酸、色氨酸、脯氨酸、丝氨酸、苏氨酸、天冬酰胺、谷氨酰胺、天冬氨酸、谷氨酸、组氨酸、赖氨酸或精氨酸组成的20种天然存在的氨基酸。例如,使用
r6=赖氨酸的氨基酰基的式ia的化合物的结构如下:
[0152][0153]
每个r7独立地是氢、低级烷基、苯基、取代的苯基、低级烷基苯基、-ch2(chor8)
m-ch2or8;2-呋喃基或3-呋喃基;
[0154]
在优选实施方案中,r7是异丙基、乙基或2-呋喃基。
[0155]
在特别优选的实施方案中,r7是h或甲基。
[0156]
本发明化合物中的选定取代基以一定的递归程度呈现。在这种情况下,“递归取代基”意指取代基可引述其本身的另一个实例。由于此类取代基的递归性质,理论上,在任何给定的实施方案中可能存在大量的化合物。例如,r9含有r
13
取代基。r
13
可以含有r
10
取代基,r
10
可以含有r9取代基。药物化学领域的普通技术人员理解,此类取代基的总数合理地受所希望的化合物的期望性质限制。此类性质包括例如但不限于物理性质诸如分子量、溶解度或log p,应用性质诸如针对目标靶的活性,以及实用性质诸如易合成性等。
[0157]
作为实例而非限制,在某些实施方案中,r5、r
13
和r
10
是递归取代基。通常,这些取代基中的每一个在给定的实施方案中可以独立地出现20、19、18、17、16、15、14、13、12、11、10、9、8、7、6、5、4、3、2、1或0次。更典型地,在给定的实施方案中,这些取代基中的每一个可以独立地出现12次或更少次。更典型地,在给定的实施方案中,r9将出现0至8次,在给定的实施方案中,r
13
将出现0至6次,在给定的实施方案中,r
10
将出现0至6次。甚至更典型地,在给定的实施方案中,r9将出现0至6次,在给定的实施方案中,r
13
将出现0至4次,在给定的实施方式中r
10
将出现0至4次。
[0158]
递归取代基是本发明的预期方面。药物化学领域的普通技术人员理解这些取代基的通用性。本发明的一个实施方案中存在递归取代基的程度,将如上所述确定总数。
[0159]
每个-het-独立地是-n(r7)-、-n(r
10
)-、-s-、-so-、-so
2-;-o-、-so2nh-、-nhso
2-、-nr7co-、-conr
7-、-n(r
13
)-、-so2nr
13-、-nr
13
co-或-conr
13-。在优选实施方案中,-het-是-o-、-n(r7)-或-n(r
10
)-。最优选地,-het-是-o-。
[0160]
每个-连接基-独立地是-o-、-(ch2)
n-、-o(ch2)
m-、-nr
13-c(=o)-nr
13-、-nr
13-c(=o)-(ch2)
m-、-c(=o)nr
13-(ch2)
m-、-(ch2)
n-(z)
g-(ch2)
n-、-s-、-so-、-so
2-、-so2nr
7-、-so2nr
10-或-het-。在优选实施方案中,-连接基-是-o-、-(ch2)
n-、-nr
13-c(=o)-(ch2)
m-或-c(=o)nr
13-(ch2)
m-。
[0161]
每个-cap是每个cap,独立地是
[0162]
每个cap独立地是
[0163]
[0164]
[0165][0166]
在优选实施方案中,cap是
[0167][0168]
每个g独立地是1至6的整数。因此,每个g可以是1、2、3、4、5或6。
[0169]
每个m独立地是1至7的整数。因此,每个m可以是1、2、3、4、5、6或7。
[0170]
每个n独立地是0至7的整数。因此,每个m可以是0、1、2、3、4、5、6或7。
[0171]
每个z独立地是-(choh)-、-c(=o)-、-(chnr7r
10
)-、-(c=nr
10
)-、-nr
10-、-(ch2)
n-、-(chnr
13
r
13
)-、-(c=nr
13
)-、o-nr
13-或co2h。如在某些实施方案中由(z)
g
指定的,z可出现1、2、3、4、5或6次,并且z的每次出现独立地是-(choh)-、-c(=o)-、-(chnr7r
10
)-、-(c=nr
10
)-、-nr
10-、-(ch2)
n-、-(chnr
13
r
13
)-、-(c=nr
13
)-或-nr
13-。因此,通过举例的方式而非限制性的方式,(z)
g
可以是-(choh)-(chnr7r
10
)-、-(choh)-(chnr7r
10
)-c(=o)-、-(choh)-(chnr7r
10
)-c(=o)-(ch2)
n-、-(choh)-(chnr7r
10
)-c(=o)-(ch2)
n-(chnr
13
r
13
)-、-(choh)-(chnr7r
10
)-c(=o)-(ch2)
n-(chnr
13
r
13
)-c(=o)-等。
[0172]
在含有-chor
8-或-ch2or8基团的任何变量中,当任何-chor
8-或-ch2or8基团相对于彼此位于1,2-或1,3-时,r8基团可任选地一起用于形成环状单取代或二取代的1,3-二噁烷或1,3-二氧戊环。
[0173]
可制备本文所述的化合物并将其用作游离碱。或者,可制备所述化合物并将其以药学上可接受的盐使用。药学上可接受的盐是保留或增强母体化合物的所需生物活性并且不产生不希望的毒理学作用的盐。此类盐的实例是(a)与无机酸例如盐酸、氢溴酸、硫酸、磷酸、硝酸等形成的酸加成盐;(b)与有机酸诸如例如乙酸、草酸、酒石酸、琥珀酸、马来酸、富马酸、葡糖酸、柠檬酸、苹果酸、抗坏血酸、苯甲酸、鞣酸、棕榈酸、藻酸、聚谷氨酸、萘磺酸、甲磺酸、对-甲苯磺酸、萘二磺酸、聚半乳糖醛酸、丙二酸、磺基水杨酸、乙醇酸、2-羟基-3-萘甲酸盐、双羟萘酸盐、水杨酸、硬脂酸、苯二甲酸、扁桃酸、乳酸等形成的盐;和(c)从元素阴离子例如氯、溴和碘形成的盐。
[0174]
需要注意的是,本发明包括式i(ia-id)范围内的化合物的所有对映异构体、非对映异构体和外消旋混合物、互变异构体、多晶型物、假多晶型物和药学上可接受的盐。此类对映异构体和非对映异构体的所有混合物都在本发明的范围内。
[0175]
式i的化合物及其药学上可接受的盐可以作为不同的多晶型物或假多晶型物存在。如本文中所用,晶体多晶型意指晶体化合物以不同晶体结构存在的能力。晶体多晶型可由晶体堆积(堆积多晶型)的差异或相同分子的不同构象异构体(构象多晶型)之间的堆积差异导致。如本文中所用,晶体假多晶型意指化合物的水合物或溶剂化物以不同晶体结构
存在的能力。本发明的假多晶型物可由于晶体堆积(堆积假多晶型)的差异或由于相同分子的不同构象异构体(构象假多晶型物)之间的堆积的差异而存在。本发明包括式i-iii的化合物的所有多晶型物和假多晶型物及其药学上可接受的盐。
[0176]
式i的化合物及其药学上可接受的盐也可以以无定形固体形式存在。如本文中所用,无定形固体是一种固体,其中在该固体中原子的位置不存在长程有序性。当晶体尺寸为2纳米或更小时,该定义也适用。可以使用包括溶剂在内的添加剂来产生本发明的无定形形式。本发明包括式i-iii的化合物的所有无定形形式及其药学上可接受的盐。
[0177]
式i的化合物可以以不同的互变异构形式存在。本领域技术人员将认识到,脒、酰胺、胍、脲、硫脲、杂环等可以以互变异构形式存在。式i的所有实施方案的脒、酰胺、胍、脲、硫脲、杂环等的所有可能的互变异构形式都在本发明的范围内。
[0178]“对映异构体”是指化合物的两种立体异构体,它们是彼此不重叠的镜像。
[0179]
本文使用的立体化学定义和约定通常遵循s.p.parker,编辑,mcgraw-hill dictionary of chemical terms(1984)mcgraw-hill book company,new york;以及eliel,e.和wilen,s.,stereochemistry of organic compounds(1994)john wiley&sons,inc.,new york。许多有机化合物以光学活性形式存在,即它们具有旋转平面偏振光的平面的能力。在描述光学活性化合物中,前缀d和l或r和s用于表示分子围绕其手性中心的绝对构型。前缀d和l、d和l或(+)和(-)用于表示通过化合物产生的平面偏振光的旋转符号,s、(-)或l表示化合物是左旋的,而以r、(+)或d为前缀的化合物是右旋的。对于给定的化学结构,除了它们是彼此的镜像外,这些立体异构体是相同的。具体的立体异构体也可称为对映异构体,此类异构体的混合物通常称为对映异构体混合物。对映异构体的50:50混合物称为外消旋混合物或外消旋物,其可在化学反应或过程中不存在立体选择性或立体定向性的情况下发生。术语“外消旋混合物”和“外消旋物”是指没有光学活性的两种对映异构体种类的等摩尔混合物。
[0180]
基本上不含其立体异构体的单一立体异构体,例如对映异构体可通过使用方法(诸如使用光学活性拆分剂形成非对映异构体)拆分外消旋混合物来获得("stereochemistry of carbon compounds,"(1962)by e.l.eliel,mcgraw hill;lochmuller,c.h.,(1975)j.chromatogr.,113:(3)283-302)。本发明的手性化合物的外消旋混合物可以通过任何合适的方法分开和分离,所述方法包括:(1)用手性化合物形成离子型非对映异构体盐,通过分级结晶或其它方法进行分离,(2)用手性衍生剂形成非对映异构体化合物,分离非对映异构体,并转化成纯立体异构体,和(3)在手性条件下直接分离基本上纯的或富集的立体异构体。
[0181]“非对映异构体”是指具有两个或更多个手性中心的立体异构体,其分子不是彼此的镜像。非对映异构体具有不同的物理性质,例如熔点、沸点、光谱性质和反应性。非对映异构体的混合物可以在高分辨率分析方法诸如电泳和色谱下分离。
[0182]
不限于任何特定理论,据信式(i)、式ii或式iii的化合物在体内具有生物还原功能。通过阻断存在于粘膜表面中的上皮钠通道,式(i)、式ii或式iii的化合物减少粘膜表面的水分吸收。这种作用在粘膜表面上增加了保护性液体的体积,重新平衡了系统,从而治疗了疾病。
[0183]
可制备本文所述的化合物并将其用作游离碱。或者,可制备所述化合物并将其以
药学上可接受的盐使用。药学上可接受的盐是保留或增强母体化合物的所需生物活性并且不产生不希望的毒理学作用的盐。此类盐的实例是(a)与无机酸例如盐酸、氢溴酸、硫酸、磷酸、硝酸等形成的酸加成盐;(b)与有机酸诸如例如乙酸、草酸、酒石酸、琥珀酸、马来酸、富马酸、葡糖酸、柠檬酸、苹果酸、抗坏血酸、苯甲酸、鞣酸、棕榈酸、藻酸、聚谷氨酸、萘磺酸、甲磺酸、对-甲苯磺酸、萘二磺酸、聚半乳糖醛酸、丙二酸、磺基水杨酸、乙醇酸、2-羟基-3-萘甲酸盐、双羟萘酸盐、水杨酸、硬脂酸、苯二甲酸、扁桃酸、乳酸等形成的盐;和(c)从元素阴离子例如氯、溴和碘形成的盐。
[0184]
需要注意的是,在式(x范围内的化合物的所有对映异构体、非对映异构体和外消旋混合物、互变异构体、多晶型物、假多晶型物和药学上可接受的盐均包括在本发明中。此类对映异构体和非对映异构体的所有混合物都在本发明的范围内。
[0185]
式i的化合物及其药学上可接受的盐可以作为不同的多晶型物或假多晶型物存在。如本文中所用,晶体多晶型意指晶体化合物以不同晶体结构存在的能力。晶体多晶型可由晶体堆积(堆积多晶型)的差异或相同分子的不同构象异构体(构象多晶型)之间的堆积差异导致。如本文中所用,晶体假多晶型意指化合物的水合物或溶剂化物以不同晶体结构存在的能力。本发明的假多晶型物可由于晶体堆积(堆积假多晶型)的差异或由于相同分子的不同构象异构体(构象假多晶型物)之间的堆积的差异而存在。本发明包括式i-iii的化合物的所有多晶型物和假多晶型物及其药学上可接受的盐。
[0186]
式i的化合物及其药学上可接受的盐也可以以无定形固体形式存在。如本文中所用,无定形固体是一种固体,其中在该固体中原子的位置不存在长程有序性。当晶体尺寸为2纳米或更小时,该定义也适用。可以使用包括溶剂在内的添加剂来产生本发明的无定形形式。本发明包括式i的化合物的所有无定形形式及其药学上可接受的盐。
[0187]
式i的化合物可以以不同的互变异构形式存在。本领域技术人员将认识到,脒、酰胺、胍、脲、硫脲、杂环等可以以互变异构形式存在。式i-iv的所有实施方案的脒、酰胺、胍、脲、硫脲、杂环等的所有可能的互变异构形式都在本发明的范围内。
[0188]“对映异构体”是指化合物的两种立体异构体,它们是彼此不重叠的镜像。
[0189]
本文使用的立体化学定义和约定通常遵循s.p.parker,编辑,mcgraw-hill dictionary of chemical terms(1984)mcgraw-hill book company,new york;以及eliel,e.和wilen,s.,stereochemistry of organic compounds(1994)john wiley&sons,inc.,new york。许多有机化合物以光学活性形式存在,即它们具有旋转平面偏振光的平面的能力。在描述光学活性化合物中,前缀d和l或r和s用于表示分子围绕其手性中心的绝对构型。前缀d和l、d和l或(+)和(-)用于表示通过化合物产生的平面偏振光的旋转的符号,s、(-)或l表示化合物是左旋的,而以r、(+)或d为前缀的化合物是右旋的。对于给定的化学结构,除了它们是彼此的镜像外,这些立体异构体是相同的。具体的立体异构体也可称为对映异构体,此类异构体的混合物通常称为对映异构体混合物。对映异构体的50:50混合物称为外消旋混合物或外消旋物,其可在化学反应或过程中不存在立体选择性或立体定向性的情况下发生。术语“外消旋混合物”和“外消旋物”是指没有光学活性的两种对映异构体种类的等摩尔混合物。
[0190]
基本上不含其立体异构体的单一立体异构体,例如对映异构体可通过使用方法(诸如使用光学活性拆分剂形成非对映异构体)拆分外消旋混合物来获得("
stereochemistry of carbon compounds,"(1962)by e.l.eliel,mcgraw hill;lochmuller,c.h.,(1975)j.chromatogr.,113:(3)283-302)。本发明的手性化合物的外消旋混合物可以通过任何合适的方法分开和分离,所述方法包括:(1)用手性化合物形成离子型非对映异构体盐,和通过分级结晶或其它方法分离,(2)用手性衍生剂形成非对映异构体化合物并分离非对映异构体,并转化成纯立体异构体,和(3)在手性条件下直接分离基本上纯的或富集的立体异构体。
[0191]“非对映异构体”是指具有两个或更多个手性中心的立体异构体,其分子不是彼此的镜像。非对映异构体具有不同的物理性质,例如熔点、沸点、光谱性质和反应性。非对映异构体的混合物可以在高拆分分析方法诸如电泳和色谱下分离。
[0192]
如上所述,用于制备本发明组合物的化合物可以呈药学上可接受的游离碱的形式。由于化合物的游离碱通常比盐在水性溶液中更不可溶,因此游离碱组合物用来提供至肺中的更持续释放的活性剂。未溶解于溶液中的以颗粒形式存在于肺中的活性剂不能诱导生理反应,而是用作逐渐溶解于溶液中的生物可用药物的贮库。
[0193]
在优选实施方案中,式(i)的化合物是
[0194][0195]
在另一个优选实施方案中,式(i)的化合物是
[0196][0197]
在另一个优选实施方案中,式(i)的化合物是
[0198][0199]
在另一个优选实施方案中,式(i)的化合物是
[0200][0201]
在另一个优选实施方案中,式(i)的化合物是
[0202][0203]
在另一个优选实施方案中,式(i)的化合物是
[0204][0205]
在另一个优选实施方案中,式(i)的化合物是
[0206][0207]
在另一个优选实施方案中,式(i)的化合物是
[0208][0209]
在另一个优选实施方案中,式(i)的化合物是
[0210][0211]
在另一个优选实施方案中,式(i)的化合物是
[0212][0213]
在另一个优选实施方案中,式(i)的化合物是
[0214][0215]
在另一个优选实施方案中,式(i)的化合物是
[0216][0217]
在另一个优选实施方案中,式(i)的化合物是
[0218][0219]
在另一个优选实施方案中,式(i)的化合物是
[0220][0221]
在另一个优选实施方案中,式(i)的化合物是
[0222][0223]
在另一个优选实施方案中,式(i)的化合物是
[0224][0225]
在另一个优选实施方案中,式(i)的化合物是
[0226][0227]
在另一个优选实施方案中,式(i)的化合物是
[0228][0229]
在另一个优选实施方案中,式(i)的化合物是
[0230]
[0231]
在特别优选的实施方案中,式(i)的化合物是
[0232][0233]
在另一个特别优选的实施方案中,式(i)的化合物是
[0234][0235]
在另一个特别优选的实施方案中,式(i)的化合物是
[0236][0237]
在另一个特别优选的实施方案中,式(i)的化合物是
[0238][0239]
单硫醇粘液溶解药的前药:
[0240]
许多现代药物是通过高通量筛选或组合化学发现的。通常就这些化合物的高药理功效选择所述化合物,但所述化合物无意中具有差的药物样特征(例如,溶解度、生物利用度、稳定性)。克服这些物理化学、生物药物学和药代动力学限制的一个策略是使用所述化合物的前药形式,即在体内经历酶促或化学转化之前无活性的分子。取决于修饰的类型,前药可具有优于其活性对应物的关键有利方面:(1)增加的稳定性和架存期,(2)增加的水溶性,(3)改善的生物利用度,(4)增加的亲脂性/通透性,和(5)改善的肠胃外给药。
[0241]
在全世界批准的药物中,5-7%可以归类为前药。这些药物分为两类,即生物前体前药或载体连接基的前药。生物前体前药通过代谢或化学转变转化为药理活性药物。载体连接基的前药具有与活性母体分子共价连接的前体部分。通常通过酶促水解释放该前体部分,该前体部分一旦被递送至治疗位置就激活母体分子。前药部分的设计通常基于需要在特定分子中改进的药物样特征、可适于前体部分的可用官能团和靶向器官或组织。在其中由于诸如空间位阻等原因而不能直接附接前体部分的情况下,也可添加间隔基或连接基。
为了耐受良好,在到达治疗组织之前,前体部分应当是非免疫原性的,稳定的,并且一旦从母体裂解出来,被迅速地从体内排泄。由于酯类易于通过普遍存在的酯酶(例如,乙酰胆碱酯酶、丁酰胆碱酯酶、羧酸酯酶、胆固醇酶)从母体药物中除去,通过掩蔽电荷基团诸如羧酸和磷酸盐,能够增加药物溶解度,以及相对简单地合成,因此它们是最常使用的前体部分之一。用作前体部分的一些其它常用官能团是:碳酸盐、氨基甲酸盐、酰胺、磷酸盐和肟。
[0242]
前药可以特别适合用作粘膜阻塞性呼吸系统疾病诸如慢性支气管炎(cb)包括慢性支气管炎的最常见致死遗传形式囊性纤维化(cf)的吸入治疗剂。我们还假设额外的分子特征可提高单硫醇粘液溶解剂的耐受性和作用持续时间。
[0243]
具体来说,我们通过酶促整合不稳定的巯基封端基团开发了粘液溶解前药。这些前药粘液溶解剂的优点在于:1)它们是完全无活性的,因此可以防止溶液中的自动氧化;2)硫醇保护基使化合物完全无臭;和3)分子可被设计来改变体内活化速率,因此可用于减缓化合物活化并延长药理作用的持续时间。
[0244]
本发明提供了一系列通过存在于细胞外环境中的常见酶(例如,核苷酸酶、磷酸酯酶和酯酶)活化的粘液溶解前药(参见r6=除h以外的物质)。作为概念验证,测试了在活化酯酶存在或不存在的情况下,前药化合物46的还原动力学。在测试条件下,单独的46不还原二硫键,
[0245][0246]
而母体分子47在<10秒内完全还原所有可用的二硫化物。然而,加入能够酶促46的酶产生浓度依赖性增加反应速率。重要的是,p-46和47都可以还原人粘液样品中的muc5b,动力学与上文中预测的相似,表明活化所需的酶活性存在于粘液中(图8和9)。
[0247]
如上所述,用于制备本发明组合物的化合物可以是药学上可接受的游离碱或酸加成盐的形式。在优选实施方案中,式(i)的前药化合物是
[0248][0249]
在另一个优选实施方案中,式(i)的前药化合物是
[0250]
[0251]
在另一个优选实施方案中,式(i)的前药化合物是
[0252][0253]
在另一个优选实施方案中,式(i)的前药化合物是
[0254][0255]
在另一个优选实施方案中,式(i)的前药化合物是
[0256][0257]
在另一个优选实施方案中,式(i)的前药化合物是
[0258][0259]
在另一个优选实施方案中,式(i)的前药化合物是
[0260][0261]
在另一个优选实施方案中,式(i)的前药化合物是
[0262][0263]
在另一个优选实施方案中,式(i)的前药化合物是
[0264][0265]
在另一个优选实施方案中,式(i)的前药化合物是
[0266][0267]
在另一个优选实施方案中,式(i)的前药化合物是
[0268][0269]
在另一个优选实施方案中,式(i)的前药化合物是
[0270][0271]
在另一个优选实施方案中,式(i)的前药化合物是
[0272][0273]
在另一个优选实施方案中,式(i)的前药化合物是
[0274][0275]
在另一个优选实施方案中,式(i)的前药化合物是
[0276][0277]
在另一个优选实施方案中,式(i)的前药化合物是
[0278][0279]
在另一个优选实施方案中,式(i)的前药化合物是
[0280][0281]
在另一个优选实施方案中,式(i)的前药化合物是
[0282][0283]
在另一个优选实施方案中,式(i)的前药化合物是
[0284][0285]
在另一个优选实施方案中,式(i)的前药化合物是
[0286][0287]
在另一个优选实施方案中,式(i)的前药化合物是
[0288][0289]
在特别优选的实施方案中,式(i)的前药化合物是
[0290][0291]
在另一个特别优选的实施方案中,式(i)的前药化合物是
[0292][0293]
在另一个特别优选的实施方案中,式(i)的前药化合物是
[0294][0295]
在另一个特别优选的实施方案中,式(i)的前药化合物是
[0296][0297]
本发明还提供了利用如上所述的本文所述的化合物的性质的治疗方法。因此,可以通过本发明的方法治疗的受试者包括但不限于患有囊性纤维化、肺纤维化、哮喘、原发性纤毛运动障碍、慢性支气管炎、支气管扩张慢性阻塞性气道疾病的患者、人工通气患者、急性肺炎患者等。本发明可用于通过向患者的至少一个肺施用活性化合物,然后从该患者诱导或收集痰样品来从患者获得痰样品。通常,通过液体气雾剂、干粉或灌洗剂将本发明施用于呼吸道粘膜表面。
[0298]
可通过本发明的方法治疗的受试者还包括鼻内施用补充氧(倾向于干燥气道表面的方案)的患者;患有影响鼻气道表面的过敏性疾病或反应(例如,对花粉、灰尘、动物毛发或颗粒、昆虫或昆虫颗粒等的过敏反应)的患者;患有鼻气道表面的细菌感染,例如葡萄球菌属(staphylococcus)感染诸如金黄色葡萄球菌(staphylococcus aureus)感染、流感嗜血杆菌(hemophilus influenza)感染、肺炎链球菌(streptococcus pneumoniae)感染、恶臭假单胞菌(pseudomonas aeuriginosa)感染等的患者;患有影响鼻气道表面的炎性疾病的患者;或患有窦炎(其中通过施用促进窦内充血流体排出的有效量来施用一种或多种活性剂以促进窦内充血粘液分泌物的排出)或组合的鼻窦炎的患者。本发明可通过局部递送(包括气雾剂和滴剂)施用于鼻窦表面。
[0299]
本发明可用于改善纤维化特发性间质性肺炎(fiip),特征在于导致显著的发病率和死亡率的肺泡间质的进行性瘢痕形成的肺部疾病。特发性肺纤维化(ipf)是最常见和最严重的fiip形式,中位生存期为3年,在美国每年影响5万个体,随着我们的人口老龄化,流行率将会增加(g.raghu,d.weycker,j.edelsberg,w.z.bradford,g.oster,incidence and prevalence of idiopathic pulmonary fibrosis.am j respir crit care med 174,810(2006年10月1日)。重要的是,大多数以前尝试的疗法已被靶向ipf中的纤维增生基质,但这些已被证明是无效的。罕见突变与ipf相关,然而,这些变体占归因危险度的一小部分。muc5b是已建立的ipf的高度重要且常见的遗传风险因子(m.a.seibold等,a common muc5b promoter polymorphism and pulmonary fibrosis.n engl j med 364,1503(2011年4月21日))的最近发现表明muc5b变体具有检测临床前疾病或轻度疾病的潜力。因此,本发明可用于治疗肺纤维化中的muc5b的潜在缺陷。
[0300]
本发明可用于改善除气道表面外的粘液清除。这样的其它粘膜表面包括胃肠表面、口腔表面、生殖-尿道表面以及眼睛表面或眼的表面。例如,可以以有效量通过任何合适的方式包括局域/局部、口服或经直肠施用本发明的活性化合物。
[0301]
在另一方面,提供了用于治疗来自空气传播病原体的感染的暴露后预防性治疗或治疗性治疗,包括向有抗来自空气传播病原体的感染这样治疗需求的个体的肺部施用有效量的式(i-vii)的化合物。可通过本发明的暴露后预防性、抢救性和治疗性治疗方法防止的病原体包括可通过口、鼻或鼻气道进入身体,从而进入肺部的任何病原体。通常,病原体将
是空气传播的病原体,其是天然存在的或通过烟雾化产生的。病原体可能是天然存在的,或者可能已在烟气雾化或将病原体引入环境中的其它方法之后被有意地引入环境中。在空气中非天然传播的许多病原体已经或可能被雾化以用于生物恐怖。对于其本发明治疗可能是有用的病原体包括但不限于niaid提出的a、b和c类优先病原体。这些类别一般对应于由疾病控制和预防中心(cdc)编制的列表。根据cdc的规定,a类试剂是可轻易地在人与人之间扩散或传播,造成高死亡率,具有造成重大公共健康影响的潜力的那些试剂。b类试剂是下一优先的,包括中度易于传播,引起中度发病率和低死亡率低的那些试剂。c类由新出现的病原体组成,所述病原体由于其可用性,产生和传播的容易性以及造成高发病率和死亡率的潜力,而在未来可被工程化以用于大规模传播。这些病原体的具体例子是炭疽和瘟疫。可被抵御的或源自其的感染风险降低的其它病原体包括流感病毒、鼻病毒、腺病毒和呼吸道合胞病毒等。可被抵御的另外的病原体是被认为引起严重急性呼吸综合征(sars)的冠状病毒。
[0302]
本发明还涉及式i的粘液溶解剂或其药学上可接受的盐用于预防、减轻和/或治疗由对放射性物质,特别是来自核袭击诸如放射性散发性装置(rdd)的引爆或事故诸如核电厂灾难等的含放射性核素的可吸入气雾剂的暴露引起的对呼吸道的确定性健康影响的用途。因此,本文提供了用于预防、减轻和/或治疗有此需要的受体中包括有此需要的人中的由含有放射性核素的可吸入的气雾剂引起的对呼吸道和/或其它身体器官的确定性健康影响的方法,所述方法包括向所述人施用有效量的式(i)的化合物或其药学上可接受的盐。
[0303]
与计划用于公众成员对来自核袭击诸如放射性散发性装置(rdd)的引爆或事故诸如核电厂灾难等的含放射性核素的可吸入气雾剂的暴露的后果管理相关的一个主要问题是如何防止、减轻或治疗对呼吸道,主要是肺的潜在确定性健康影响。必须要有准备的药物、技术和程序以及经过培训的人员来管理和治疗此类高度内部污染的个体。
[0304]
已进行了研究来确定通过其防止、减轻或治疗由内部沉积的放射性核素引起的对呼吸道和身体中的各种器官的潜在损害的方法。迄今为止,大多数研究注意力集中在设计成通过加速其排泄或去除来减轻内部沉积的放射性核素对健康的影响的策略。这些策略集中在能够到达血流并沉积在特异于给定的放射性元素的远端系统部位的可溶性化学形式。在其中沉积的放射性核素呈相对不溶的形式的情况下,此类方法将不起作用。研究表明,许多(如果不是大多数的话)来自rdd的分散的放射性核素的物理化学形式将呈相对不溶的形式。
[0305]
已知有效减少可吸入不溶性放射性气雾剂至肺的辐射剂量的唯一方法是支气管肺泡灌洗液或bal。从已用于治疗肺泡蛋白沉积症患者的技术改进的这种技术已被证明是安全的、可重复的方法,即使在延长的时期内进行亦如此。虽然程序上有差异,但bal的基本方法是麻醉受试者,然后将等渗盐水缓慢地引入单个肺叶,直到达到功能残气量。然后加入额外的体积并通过重力排出。
[0306]
在动物上使用bal的研究结果表明,通过合理的bal顺序可除去约40%的深肺含量。在一些研究中,动物在回收的放射性核素量方面存在相当大的变化性。目前尚不了解变化性的原因。
[0307]
此外,根据对动物的研究,据信bal疗法的显著剂量减少导致由于吸入不溶性放射性核素而造成的健康影响减轻。在研究中,成年狗吸入不溶性
144
ce-fap颗粒。两组狗被给予
已知导致放射性肺炎和肺纤维化(约2mbq/kg体重)的
144
ce肺含量,一组在暴露后2至56天用10次单侧灌洗液处理,另一组未经处理。将第三组暴露于可与治疗后在bal处理组中看到的相当的
144
ce水平(约1mbq/kg),但是这些动物未经处理。使所有动物都按其寿命活着,所述寿命延长至16年。由于每组中狗之间的
144
ce的初始肺含量存在差异,因此每组的剂量率和累积剂量重叠。然而,从存活曲线来看,bal在降低肺炎/纤维化风险方面的作用是显而易见的。在肺含量为1.5-2.5mbq/kg的未处理的狗中,平均存活时间为370
±
65天。对于经处理的狗,平均存活时间为1270
±
240天,差异在统计上是显著的。接受0.6-1.4mbq的
144
ce的肺活量的第三组具有1800
±
230天的平均存活时间,这与处理组无统计学差异。对于增加的生存率,同样重要的是,高剂量未处理组中的狗死于对肺的确定性影响(肺炎/纤维化),而经处理的狗不死于对肺的确定性影响。相反,经处理的狗,如低剂量未治疗组中的狗,主要有肺肿瘤(血管肉瘤或癌)。因此,bal治疗引起的剂量减少似乎已经在肺中产生了基于肺接受的辐射剂量可预测的生物学效应。
[0308]
基于这些结果,有人认为通过用于增强从肺的颗粒清除的任何方法或其组合进一步降低残留放射剂剂量将进一步降低对肺的健康影响的可能性。然而,bal是具有许多缺点的过程。bal是高度侵入性的过程,必须由专业的医疗中心由训练有素的肺科医师进行。因此,bal过程是昂贵的。鉴于bal的缺点,对于需要加速去除放射性颗粒的人员来说,例如在发生核袭击的情况下,这不是容易和立即可用的治疗选择。在发生核袭击或核事故的情况下,需要对已经暴露或有暴露危险的人立即和相对容易地进行治疗。已显示作为吸入气雾剂施用的钠通道阻滞剂恢复气道表面的水合。气道表面的这种水合有助于从肺中清除积聚的粘液分泌物和相关的颗粒物质。因此,不受任何特定理论的约束,据信钠通道阻断剂可以与本发明中所述的粘液溶解剂组合使用,以加速从气道通道中除去放射性颗粒。
[0309]
如上所述,放射性袭击诸脏弹后肺部的最大风险由吸入和保留不溶性放射性颗粒所致。作为放射性颗粒保留的结果,对肺的累积暴露显著增加,最终导致肺纤维化/肺炎和潜在的死亡。不溶性颗粒不能被螯合剂系统地清除,因为这些颗粒不在溶液中。迄今为止,通过bal物理去除颗粒物是经显示在减轻放射诱导的肺部疾病中是有效的唯一的治疗方案。如上所述,bal不是用于减轻已被吸入体内的放射性颗粒的影响的现实治疗方案。因此,期望提供有效地帮助从气道通道清除放射性颗粒的治疗方案,并且与bal不同,所述方案在大规模的辐射照射情景中可相对简单地进行施用并且是可扩展的。另外,还希望治疗方案在相当短的时期内可容易地被许多人使用。
[0310]
在本发明的一个方面,用于预防、减轻和/或治疗由含有放射性核素的可吸入气雾剂引起的对呼吸道和/或其它身体器官的确定性健康影响的方法包括向有此需要的个体施用有效量的式i的粘液溶解剂或其药学上可接受的盐。在该方面的特征中,将粘液溶解剂与渗透物一起施用。进一步考虑到该特征,渗透物是高渗盐水。在另一个特征中,将粘液溶解剂和渗透物与离子转运调节剂结合施用。进一步考虑到该特征,离子转运调节剂可选自β-激动剂、cftr增效剂、嘌呤能受体激动剂、鲁比前列酮和蛋白酶抑制剂。在该方面的另一个特征中,放射性核素选自钴-60、铯-137、铱-192、镭-226、磷-32、锶-89和90、碘-125、铊-201、铅-210、钍-234、铀-238、钚、钴-58、铬-51、镅和锔。在另一个特征中,放射性核素来自放射性处置装置。在另一个特征中,可将粘液溶解剂或其药学上可接受的盐在个体吸入的可吸入颗粒的气雾剂悬浮液中施用。在另外的特征中,在暴露于放射性核素之后施用粘液
溶解剂或其药学上可接受的盐。
[0311]
本发明主要涉及人受试者的治疗,但也可出于兽医目的用于治疗其它哺乳动物受试者,诸如狗和猫。
[0312]
本发明的另一方面是在药学上可接受的载体(例如,水性载体溶液)中包含式i的化合物的药物组合物。通常,以有效降低粘膜表面上粘液粘度的量将式i的化合物包含在组合物中。
[0313]
粘液溶解剂联合疗法:
[0314]
本发明的一个方面是粘液溶解剂与其它药物或赋形剂的组合,以改善本发明所述化合物的功效和耐受性。
[0315]
在本发明的一个实施方案中,利用粘液溶解剂来为其它治疗剂提供通过粘液层至气道上皮的通路。粘液形成扩散屏障,其可阻止治疗分子到达它们预期的作用位点。
[0316]
通过利用本发明所述的粘液溶解剂的预处理或与其的共处理,可增强以下治疗剂至其在呼吸道上皮中的作用位点的到达。
[0317]
在本发明的一个实施方案中,将粘液溶解剂与其它疗法或小分子药物组合使用以提供有利的添加剂或协同益处。
[0318]
粘液溶解剂与渗透活性物的组合:
[0319]
本发明的另一方面是将还原剂与渗透物组合施用。恢复具有粘膜阻塞性疾病的受试者的气道表面水合的简单方法是吸入高渗渗透物溶液(最常见地是7%高渗盐水(hs)),其将水吸入气道表面。气道表面的润滑剂纤周层(pcl)的再水合有利于粘液清除,并因此除去吸入的感染因子。通过施用高渗溶液获得的粘液再水合和通过还原剂破坏粘液聚合物预期会产生粘液粘度和弹性的协同降低,从而改善粘液的可运输性和清除率。
[0320]
吸入hs是一种独特的治疗剂,因为其被全国~60%的cf患者使用,但未被fda批准用于肺部疾病的日常使用。因此,hs尚未经历严格的临床测试,以确定最有效并且耐受性良好的剂量和给药频率。相反,hs方案在患者和医生的实践中得到了优化。最常见的是,将hs以每次治疗使用4ml 7%的高渗盐水两次15分钟的吸入治疗进行施用。患者使用的hs(7%的nacl)的张力已被确定为通常被耐受的最大浓度(即最小的刺激或支气管收缩)。
[0321]
还可将式i的化合物与渗透物结合使用,从而降低水合物粘膜表面所需的化合物的剂量。这一重要性质意味着化合物将具有较低的引起不期望的副作用的倾向。本发明的活性渗透物是具有渗透活性的分子或化合物(即,为“渗透物”)。本发明的“渗透活性”化合物在气道或肺上皮表面上是不可透过膜的(即,基本上不可吸收的)。如本文中所用,术语“气道表面”和“肺表面”包括肺气道表面,诸如支气管和细支气管、肺泡表面以及鼻和窦表面。本发明的活性化合物可以是离子渗透物(即,盐),或者可以是非离子渗透物(即,糖、糖醇和有机渗透物)。特别地期望具有外消旋性质的活性化合物的两种外消旋形式包括在本发明中有用的活性化合物的组中。应当注意,本发明包括所述渗透活性化合物的所有外消旋体、对映异构体、非对映异构体、互变异构体,多晶型物和假多晶型物以及外消旋混合物。
[0322]
本发明的活性化合物可以是离子渗透物(即,盐),或者可以是非离子渗透物(即,糖、糖醇和有机渗透物)。特别地期望具有外消旋性质的活性化合物的两种外消旋形式包括在本发明中有用的活性化合物的组中。应当注意,本发明包括所述渗透活性化合物的所有外消旋体、对映异构体、非对映异构体、互变异构体、多晶型物和假多晶型物以及外消旋混
合物。
[0323]
作为离子渗透物的可用于本发明的活性渗透物包括药学上可接受的阴离子与药学上可接受的阳离子的任何盐。优选地,阴离子和阳离子之一(或两者)相对于它们所施用至的气道表面是不可吸收的(即,渗透活性且不经历快速主动转运)。此类化合物包括但不限于在fda批准的市售盐中含有的阴离子和阳离子,参见例如remington:the science and practice of pharmacy,第ii卷,第1457页(第19版1995),其通过引用并入本文,并且可以以包括其常规组合在内的任何组合使用。
[0324]
可用于实施本发明的药学上可接受的渗透活性阴离子包括但不限于乙酸根、苯磺酸根、苯甲酸根、碳酸氢根、酒石酸氢根、溴化物、依地酸钙、坎西雷特(樟脑磺酸根)、碳酸根、氯化物、柠檬酸根、二盐酸根、依地酸根、乙二磺酸根(1,2-乙烷二磺酸根)、依托酸根(月桂基硫酸根)、乙磺酸根(1,2-乙烷二磺酸根)、富马酸根、葡庚糖酸根、葡糖酸根、谷氨酸根、乙醇酰基阿散酸根(对轻乙酰氨基苯胂酸根(p-glycollamidophenylarsonate))、己基间苯二酚根、海巴明(n,n
′-
二(脱氢枞酸基)乙二胺)、氢溴酸根、盐酸根、羟基萘甲酸根、碘化物、羟乙基磺酸根、乳酸根、乳糖酸根、苹果酸根、马来酸根、扁桃酸根、甲磺酸根、甲基溴、甲基硝酸根、甲基硫酸根、粘酸根、萘磺酸根、硝酸根、硝酸根、双羟萘酸根(恩波酸根(embonate))、泛酸根、磷酸根或二磷酸根、聚半乳糖醛酸根、水杨酸根、硬脂酸根、次乙酸根、琥珀酸根、硫酸根、鞣酸根、酒石酸根、茶氯酸根(8-氯茶碱)、三乙基碘、碳酸氢根等。特别优选的阴离子包括氯化物、硫酸根、硝酸根、葡糖酸根、碘化物、碳酸氢根、溴化物和磷酸根。
[0325]
可用于实施本发明的药学上可接受的阳离子包括但不限于有机阳离子诸如苄星青霉素(n,n'-二苄基乙二胺)、氯普鲁卡因、胆碱、二乙醇胺、乙二胺、葡甲胺(n-甲基-d-葡糖胺)、普鲁卡因、d-赖氨酸、l-赖氨酸、d-精氨酸、l-精氨酸、三乙基铵、n-甲基d-甘油等。特别优选的有机阳离子是3-碳、4-碳、5-碳和6-碳有机阳离子。可用于本发明实践的金属阳离子包括但不限于铝、钙、锂、镁、钾、钠、锌、铁、铵等。特别优选的阳离子包括钠、钾、胆碱、锂、葡甲胺、d-赖氨酸、铵、镁和钙。
[0326]
可以与本文所述的钠通道阻断剂一起用于实施本发明的渗透活性盐的具体实例包括但不限于氯化钠,氯化钾,氯化胆碱,碘化胆碱,氯化锂,氯化葡甲胺,l-赖氨酸氯化物,d-赖氨酸氯化物、氯化铵,硫酸钾,硝酸钾,葡糖酸钾,碘化钾,氯化铁,氯化亚铁,溴化钾等。可以使用单一盐或不同渗透活性盐的组合来实施本发明。不同盐的组合是优选的。当使用不同的盐时,不同盐中的阴离子或阳离子之一可以相同。
[0327]
本发明的渗透活性化合物还包括非离子渗透物诸如糖、糖醇和有机渗透物。可用于本发明实践的糖和糖醇包括但不限于3-碳糖(例如甘油,二羟基丙酮);4-碳糖(例如,赤藓糖、苏糖和赤藓糖醇的d和l形式);5-碳糖(例如,核糖、阿拉伯糖、木糖、来苏糖、阿洛酮糖、果糖、山梨糖和塔格糖的d和l形式);和6-碳糖(例如,altose、阿洛糖、葡萄糖、甘露糖、古洛糖、艾杜糖、半乳糖和塔洛糖的d和l形式,以及阿洛-庚酮糖、阿洛糖-庚酮糖、葡萄-庚酮糖、甘露-庚酮糖、葡萄-庚酮糖、艾杜-庚酮糖、半乳-庚酮糖、塔洛-庚酮糖的d和l形式)。可用于本发明实施的其它糖包括棉子糖、棉子糖系列寡糖和水苏糖。可用于本发明的每种糖/糖醇的还原形式的d和l形式也是本发明范围内的活性化合物。例如,当还原时葡萄糖变成山梨糖醇;在本发明的范围内,山梨糖醇以及糖/糖醇的其它还原形式(例如,甘露糖醇、
卫矛醇、阿糖醇)是本发明的相应活性化合物。
[0328]
本发明的渗透活性化合物另外包括称为“有机渗透物”的非离子渗透物的家族。术语“有机渗透物”通常用于指用于控制肾脏中的细胞内渗透压的分子。参见例如j.s.handler等,comp.biochem.physiol,117,301-306(1997);m.burg,am.j.physiol.268,f983-f996(1995),其各自通过引用并入本文。虽然本发明人不希望受本发明的任何特定理论的约束,但这些有机渗透物似乎可用于控制气道/肺表面的细胞外体积。可在本发明中用作活性化合物的有机渗透物包括但不限于三种主要类型的化合物:多元醇(多元醇(polyhydric alcohols))、甲胺和氨基酸。被认为可用于本发明实施的多元醇有机渗透物包括但不限于肌醇、肌-肌醇和山梨糖醇。可用于本发明的实施的甲胺有机渗透物包括但不限于胆碱、甜菜碱、肉毒碱(l-、d-和dl形式)、磷酸胆碱、溶血磷脂酰胆碱、甘油磷酰胆碱、肌酸和磷酸肌酸。本发明的氨基酸有机渗透物包括但不限于甘氨酸、丙氨酸、谷氨酰胺、谷氨酸、天冬氨酸、脯氨酸和牛磺酸的d-型和l-型。可用于本发明的实施的其它渗透物包括tihulose和肌氨酸。哺乳动物有机渗透物是优选的,人有机渗透物是最优选的。然而,某些有机渗透物物是细菌、酵母和海洋动物来源的,并且这些化合物也是本发明范围内的有用的活性化合物。
[0329]
在某些情况下,可向受试者施用渗透物前体;因此,这些化合物也可用于本发明的实施。如本文中所用,术语“渗透物前体”是指通过代谢步骤,分解代谢或合成代谢步骤,转化成渗透物的化合物。本发明的渗透物前体包括但不限于作为多元醇和甲胺的前体的葡萄糖、葡萄糖聚合物、甘油、胆碱、磷脂酰胆碱、溶血磷脂酰胆碱和无机磷酸盐。在本发明范围内的氨基酸渗透物的前体包括被水解以产生渗透物氨基酸的蛋白质、肽和聚氨基酸,以及可通过代谢步骤诸如转氨作用转化成渗透物氨基酸的代谢前体。例如,氨基酸谷氨酰胺的前体是聚-l-谷氨酰胺,谷氨酸的前体是聚-l-谷氨酸。
[0330]
粘液溶解剂与钠通道阻断剂的组合:
[0331]
通过气道上皮的协调离子转运直接调节粘膜表面的水合水平。重要的是,通过上皮钠通道(enac)的钠吸收提供了水合中的限速步骤。在具有enac的功能丧失突变的人受试者中,有“湿”气道表面和极快的粘液清除率(kerem等,n engl j med.1999jul 15;341(3):156-62)。相反,已显示通过enac的增加的钠吸收是肺部cf患者的粘液脱水和粘液栓形成的根本原因。此外,在肺中过表达enac的转基因小鼠具有导致死亡的脱水的气道表面和减少的/不存在的粘液清除(hummler等,proc natl acad sci usa.1997 oct 14;94(21):11710-5)。如从临床和实验数据预测的,enac的药物阻断可保存气道表面上的液体并增加粘液清除率(hirsh等,j pharmacol exp ther.2008;325(1):77-88)。具体实例包括但不限于:
[0332]
小分子通道阻塞剂:小分子enac阻断剂能够直接阻止钠通过enac通道孔的运输。可以通过本发明的方法施用的enac阻断剂包括但不限于:阿米洛利、苯扎米尔、非那米尔和阿米洛利类似物,如由美国专利号6,858,614、美国专利号6,858,615、美国专利号6,903,105、美国专利号6,995,160、美国专利号7,026,325、美国专利号7,030,117、美国专利号7,064,129、美国专利号7,186,833、美国专利号7,189,719、美国专利号7,192,958、美国专利号7,192,959、美国专利号7,241,766、美国专利号7,247,636、美国专利号7,247,637、美国专利号7,317,013、美国专利号7,332,496、美国专利号7,345,044、美国专利号7,368,447、
美国专利号7,368,450、美国专利号7,368,451、美国专利号7,375,107、美国专利号7,399,766、美国专利号7,410,968、美国专利号7,820,678、美国专利号7,842,697、美国专利号7,868,010、美国专利号7,875,619。美国专利7,956,059、美国专利8,008,494、美国专利8,022,210、美国专利8,124,607、美国专利8,143,256、美国专利8,163,758、美国专利8,198,286、美国专利8,211,895、美国专利8,324,218、美国专利8,507,497、美国专利8,575,176、美国专利8,669,262、美国专利7,956,059、美国专利8,008,494、美国专利8,022,210、美国专利8,124,607、美国专利8,143,256、美国专利8,163,758、美国专利8,198,286、美国专利8,211,895、美国专利8,324,218、美国专利8,507,497、美国专利8,575,176、美国专利8,669,262、美国专利7,956,059、美国专利8,008,494、美国专利8,846,688、美国专利8,022,210、u.s.9,029,382、美国专利9,072,738、美国专利9,102,633、美国专利申请公开号us2014/0142118-a1、美国专利申请公开号us20140170244-a1和美国专利申请公开号us20140171447-a1所举例说明的。
[0333]
粘液溶解剂与蛋白酶抑制剂的组合:
[0334]
enac蛋白水解被适当地描述为通过enac增加钠的运输。蛋白酶抑制剂阻断内源性气道蛋白酶的活性,从而阻止enac的裂解和活化。裂解enac的蛋白酶包括弗林蛋白酶、安眠蛋白(meprin)、蛋白裂解酶(matriptase)、胰蛋白酶、通道相关蛋白酶(cap)和嗜中性粒细胞弹性蛋白酶。可通过本发明方法施用的可抑制这些蛋白酶的蛋白水解活性的蛋白酶抑制剂包括但不限于卡莫司他、前列腺蛋白酶、弗林蛋白酶、抑肽酶、亮抑酶肽和胰蛋白酶抑制剂。
[0335]
粘液溶解剂与核酸和小干扰rna(sirna)的组合:
[0336]
任何合适的核酸(或多核酸)可用于实施本发明,包括但不限于反义寡核苷酸、sirna、mirna、mirna模拟物、antagomir、核酶、适体和诱饵寡核苷酸核酸。参见,例如,美国专利申请公开号20100316628。通常,此类核酸的长度可为17个或19个核苷酸,长度达23个、25个或27个核苷酸,或更多个核苷酸。
[0337]
可使用任何合适的sirna活性剂来实施本发明。实例包括但不限于美国专利号7,517,865和美国专利申请号20100215588、20100316628、20110008366和20110104255中描述的那些sirna活性剂。通常,sirna的长度为17个或19个核苷酸,长度达23个、25个或27个核苷酸,或更多个核苷酸。
[0338]
粘液溶解剂与促分泌剂的组合:
[0339]
囊性纤维化(cf)基因的突变导致跨呼吸上皮的异常离子转运(matsui等,cell 1998;95:1005-15)。在cf患者中钠的过量吸收以及无法通过气道上皮分泌氯化物驱动水吸收下降到通过不适当的盐吸收所产生的渗透梯度,使气道粘液分泌物脱水并降低pcl中液体的体积。在copd中,香烟烟雾损害cftr功能,从而产生与cf相似的获得性表型。
[0340]
p2y2受体激动剂:可与本发明中描述的方法和分子组合施用的药剂包括一组p2y2受体激动剂。在人支气管上皮(hbe)的腔表面上富含嘌呤能(p2y2)受体,并且已知其可刺激cl-分泌并抑制na
+
吸收(goralski等,curr opin pharmacol.2010jun;10(3):294-9)。utp是内源性p2y2受体激动剂的实例,其在气道上皮中提供氯化物分泌的强烈刺激,抑制钠吸收和增加气道表面液体层,从而增加作为肺的主要防御机制的粘液清除率。使用尿苷-5-三磷酸(utp)通过气雾剂递送至cf和原发性纤维运动障碍(pcd)患者的气道表面的早期研究表
明utp在增强mc和改善平均咳嗽清除率方面的有用性。
[0341]
合适的p2y2受体激动剂描述于但不限于美国专利号6,264,975、美国专利号5,656,256、美国专利号5,292,498、美国专利号6,348,589、美国专利号6,818,629、美国专利号6,977,246、美国专利号7,223,744、美国专利号7,531,525和美国专利申请公开2009/0306009,其各自通过引用并入本文。
[0342]
替代性氯化物通道诸如cacc和clc-2类通道的活化剂:cacc在哺乳动物细胞中广泛表达,其中它们涉及广泛的生理功能,包括跨上皮液体分泌、卵母细胞受精、嗅觉和感觉信号转导、平滑肌收缩以及神经元和心脏兴奋。全细胞电流分析表明cacc亚家族之间的几个常见特征,包括膜去极化后的缓慢活化,向外整流稳态电流和比氯化物通透性大的碘化物。单通道分析提出了四种或更多种不同的cacc亚类,其广泛的报道的单通道电导从心肌细胞中小于2ps至气道上皮细胞中的50ps。
[0343]
cacc活化的后果是细胞类型特异性的,例如上皮细胞中的氯化物分泌、嗅觉受体神经元中的动作电位产生、平滑肌收缩和卵母细胞中的多精受精的防止。在一些细胞类型诸如平滑肌细胞中,膜去极化激活电压门控性钙通道,增加细胞内钙浓度。尽管近三十年前已经在功能上表征了cacc,但直到最近它们的分子身份仍然不清楚,其潜在的候选物包括bestrophins(best1-best4)(sun等,proc natl acad sci usa 99,4008-4013(2002)和tsunenari等,j biol chem 278,41114_41125(2003))、钙活化的氯化物通道clca家族蛋白质(gruber等,genomics 1998;54:200-214)和clc3(huang p等,(2001)regulation of human clc-3 channels by multifunctional ca2+/calmodulin-dependent protein kinase.jbc 276:20093-100)。三个独立的实验室已鉴定了tmem16a,也称为anoctamin1,为cacc的有力候选物(yang yd等(2008)tmem16a confers receptor-activated calcium-dependent chloride conductance.nature.455:1210-15;caputo a等(2008)tmem16a,a membrane protein associated with calcium-dependent chloride channel activity.science.322:590-4;schroeder bc等,(2008)expression cloning of tmem16a as a calcium-activated chloride channel subunit.cell.134:1019-29)。使用了三种不同的策略:对具有多个跨膜区段且未知功能的膜蛋白进行数据库搜索(yang yd等(2008)tmem16a confers receptor-activated calcium-dependent chloride conductance.nature.455:1210-15),观察到白介素4(il4)治疗的支气管上皮细胞显示增加的cacc活性之后进行功能基因组学(caputo a等(2008)tmem16a,a membrane protein associated with calcium-dependent chloride channel activity.science.322:590-4),以及使用不具有内源性cacc活性的幼体美西螈(axolotl)卵母细胞进行表达克隆(schroeder bc等(2008)expression cloning of tmem16a as a calcium-activated chloride channel subunit.cell.134:1019-29)。有确凿的证据表明tmem16a是cacc的关键组分,这些证据包括在其电生理性质方面类似于天然cacc、在多种转染的细胞系统中cacc电流的出现、在rnai敲低(knockdown)后cacc电流的减小以及其组织分布。tmem16a具有8个推定的跨膜区段,没有明显参与钙调节的结构域。
[0344]
clc2是普遍表达的、通过细胞肿胀活化的内向整流氯通道。认为clc2参与细胞体积调节,但其具有与已在许多组织中表征的体积敏感氯通道不同的生物物理学特性。在美国专利no.6015828、6159969和7253295中描述了合适的替代氯化物通道活化剂。可通过施
用本发明的化合物和方法来增强替代性氯化物通道(诸如cacc和clc-2类通道)活化剂的治疗效力。
[0345]
cftr活性的调节剂。编码cftr蛋白的基因中的突变引起遗传性致死性疾病囊性纤维化,所述cftr蛋白是在气道上皮细胞中表达的camp激活的氯化物通道。在cftr中的多种突变通过限制氯离子通过cftr向气道上皮细胞表面的分泌以及通过钠离子吸收的异常调节,引起离子运输功能障碍,从而导致钠阳离子的过量吸收。离子运输中的这些缺陷导致气道表面液体层的水合受损、粘液清除的减少以及导致肺功能的进行性丧失。最近,已显示在香烟烟雾暴露的组织中存在cftr功能缺陷,从而暗示cftr功能障碍在copd中的作用。
[0346]
已描述了cftr中1500多个推定的突变,可根据遗传缺陷的分子机制将这些突变进行分类(rowe等,pulm pharmacol ther.,23(4):268-78(2010))。每种这些突变的生物学的理解已产生基于特定突变类型的治疗策略。i类突变包括在cftr编码区内的提前终止密码子(ptc,例如,无意义突变),这会导致正常蛋白质翻译的过早截止。在10%的cf患者中发现了这些突变,但在德系犹太人(ashkenazi jews)(75%的突变型cftr等位基因)中特别常见。ii类cftr突变包括人中最常见的突变f508del cftr(占等位基因的75%,并见于约90%的cf患者中)。在508位的苯丙氨酸缺失引起cftr显示异常折叠,其特征在于核苷酸结合结构域1(nbd1)与跨膜结构域之间的结构域-结构域相互作用的稳定性不足。错误折叠的蛋白质被内质网(er)内的细胞分子伴侣所识别,送至蛋白酶体,并在到达其在细胞表面的活性位点之前被迅速降解。由于负责识别和降解错误折叠的蛋白质的细胞机制不是100%有效,特别是个体表现出f508del cftr的低水平表面表达时,这可解释在f508del cftr纯合子个体中所观察到的部分cftr活性(以及更温和的cf表型),并且可代表更易于蛋白质修复的群体。即使在细胞表面时,f508del cftr表现出降低的门控,这表明错误折叠的cftr还表现出降低的cftr离子通道活性。iii类和iv类cftr突变的特征在于全长cftr到达细胞表面,但由于异常的通道门控(iii类,例如g551d)或降低的离子通道孔的导电率(iv类,如r117h)而显示出降低的离子运输活性。类似地,剪接突变体(v类)和c末端内的突变(vi类)也是全长的,但由于质膜内活性通道的数量减少而显示出降低的活性。尽管cftr突变体的分子基础是复杂的,而且尚不完整,但cftr突变体的分类可简化为基于开发中的药剂活性的治疗相关组。常规的和高通量的药物发现程序均导致解决特定突变型cftr等位基因的新化合物的发现。这些"cftr调节剂"是旨在修复cftr蛋白质的药剂并且在以下的每个部分中进行了描述。
[0347]
细胞表面囊性纤维化跨膜电导调节子cftr突变类的增效剂导致存在于质膜上的cftr功能失调,所述cftr突变包括iii、iv、v和vi类突变并代表cftr活化剂的潜在靶标。g551d cftr表示这类药剂的原型cftr等位基因,因为其表现出正常的表面表达和半衰期,但由于核苷酸结合结构域内三磷酸腺苷(atp)结合口袋中的氨基酸取代而赋予通道门控严重的缺陷(gregory,r.j.等(1991)maturation and function of cystic fibrosis transmembrane conductance regulator variants bearing mutations in putative nucleotide-binding domains 1and 2.mcb 11:3886-93;bompadre,s.g.等(2007)g551d and g1349d,two cf-associated mutations in the signature sequences of cftr,exhibit distinct gating defects.gen physiol.129:285-298)。黄酮类是公知的突变型cftr的活化剂,并且是就其在人个体中的有益作用(包括局部施用)而被最早研究的药剂之一。虽然药剂诸如染料木素(genistein)受在鼻气道中缺少效力的影响,但是最近的努力已
经证明类黄酮槲皮素在鼻中的活性。然而,类黄酮药剂受到溶解性及全身性吸收差的挑战,而且对于吸入性疗法是差的开发候选物。更近的发现策略集中于鉴定这样的化合物:其"加强"cftr活性、恢复内源性调节(例如,环磷酸腺苷(camp)依赖的调节)和离子运输,无潜在有害的过度组成型活化(诸如见于某些腹泻疾病中的过度的cftr活化)。这种类型药剂的鉴定适合基于高通量筛选的策略以通过在基于细胞的筛选测定中测量对阴离子电导的影响而发现激活突变型cftr的药剂。许多特定的策略已用于这种类型的筛选,所述策略包括氯化物敏感染色、基于荧光共振能量的膜电位转移的分析以及气道单层的细胞电导。突变型cftr的小分子增效剂的鉴定和表征已经导致在体外和在临床中具有显著活性的药剂的开发。
[0348]
已经针对改正f508del cftr的折叠的目标做出了显著努力,从而恢复错误折叠蛋白质的离子通道活性。已开发了多样的细胞靶标,与大量目前已知与cftr生物源(biogenesis)相互作用的蛋白质相称。药剂诸如4-苯基丁酸盐下调hsc70(或其它细胞分子伴侣)关键折叠过程,并代表在临床中测试的化合物的早期实例。其它最近的努力结果来自在测试化合物与f508del表达的细胞孵育后氯化物通道功能的高通量文库筛选。许多这些策略中已经鉴定了可通过伴侣途径解决细胞生物源的f508del纠正者(corrector)。还报道这种药剂通过归因于细胞加工机械特征或降低的内吞运输的改变的表面再循环而延长f508del cftr在质膜中的半衰期的药理学活性。如果确认其在体内的安全性,则这类药剂可以是潜在的药物开发候选者。已显示其它化合物与cftr直接的相互作用并且可提供比改变细胞折叠或细胞质量控制的一般方面的药剂更高的特异性。对错误折叠蛋白质的整体细胞应答也可代表靶标。组蛋白脱乙酰酶(hdac)对基因表达具有深远的影响,并且hdac家族的特定成员参与促进f508del cftr降解的er相关降解途径。用hdac抑制剂处理cf细胞可以调节er应激,hdac诸如辛二酰苯胺异羟肟酸以及hdac的sirna沉默,提高细胞膜中f508del cftr水平。例如这些方法的组合揭示了用于f508del改正的多种潜在药剂。使用不止一种这样的策略,f508del cftr的添加剂或协同救援可提供实现足以赋予cf呼吸上皮正常表型的离子运输活性的希望。
[0349]
过早终止密码子(ptc)的通读代表了另一种解决cf以及ptc引起的许多其它遗传疾病的根本原因的令人激动的方法。某些氨基糖苷类和其它药剂具有与核糖体亚基内的真核rrna相互作用的能力。尽管这种相互作用较原核生物中所见的弱得多,并且与人个体中氨基糖苷类毒性的主要原因不同,但其可通过中断核糖体的正常纠错功能而适度降低真核生物翻译的保真度。在提前终止密码子处插入近同源氨基酸允许蛋白质翻译继续,直至达到并正确使用mrna转录物的末端上正常存在的几个终止密码子之一。该策略的特异性归因于mrna的真正末端处的更大的终止密码子保真度,并且通过说明没有可检测到的超出天然终止密码子的延伸已在体外确立。
[0350]
可与本发明中所述的方法和分子组合施用的cftr活性调节化合物包括但不限于:us 2009/0246137 a1、us 2009/0253736 a1、us 2010/0227888 a1、us 7645789、us 2009/0246820 a1、us 2009/0221597 a1、us 2010/0184739 a1、us 2010/0130547 a1、us 2010/0168094 a1、us 7553855、us 7,772,259 b2、us 7,405,233 b2、us 2009/0203752和us 7,499,570中描述的化合物。
[0351]
粘液溶解剂与抗感染剂的组合:
[0352]
慢性阻塞性肺病伴有急性和慢性细菌感染两者。急性和慢性感染均导致肺以加重形式急性发作的慢性炎症。用多种吸入式抗炎剂治疗潜在的炎症。例如,在囊性纤维化中,引起慢性感染最常见的细菌是铜绿假单胞菌(pseudomonas aeruginosa,p.aeruginosa)并且针对该细菌有效的抗生素是治疗的主要组分(flume,am j respir crit care med.176(10):957-69(2007))。同样,细菌诸如金黄色葡萄球菌(staphylococcus aureus,s.aureus)、洋葱伯克霍尔德菌(burkholderia cepacia,b.cepacia)和其它革兰阴性菌以及厌氧菌从呼吸道分泌物分离,并且患有cf的人可能从这些病原体的治疗中获益以保持他们的肺健康。厌氧细菌也被认定是cf气道、患有慢性鼻窦炎的受试者的鼻窦以及可能地患有copd的受试者的气道的特点。类似地,吸入物或微吸入物,尤其是在老年群体中和在睡眠期间,与化学性肺炎、厌氧菌感染和随后的支气管扩张相关。吸入物相关性肺炎和厌氧菌感染的理想治疗方法是即时治疗。因此,在肺部加重以及在慢性抑制治疗期间,使用抗生素根除早期感染。
[0353]
抗生素活性的主要量度是最小抑制浓度(mic)。mic是在体外完全抑制微生物生长的最低抗生素浓度。虽然mic是抗生素效力的良好指标,但其仅表示抗微生物活性的时间进程。pk参数对抗生素的肺部组织水平时间过程进行了定量。对于评价抗生素效力最重要的三个药动学参数是峰值组织水平(cmax)、波谷水平(cmin)和组织浓度时间曲线下的面积(auc)。虽然这些参数对组织水平时间过程进行了定量,但它们没有描述抗生素的杀伤活性。
[0354]
将pk参数与mic整合给予我们定量抗生素活性的三个pk/pd参数:峰值/mic比、t>mic和24小时-auc/mic比。峰值/mic比只简单地是cpmax除以mic。t>mic(高于mic的时间)是其中血清水平超过mic的剂量间隔的百分比。24小时-auc/mic比是通过24小时-auc除以mic确定的。最好地描述杀伤活性的抗生素的三种药效学性质是时间依赖性、浓度依赖性和持续效应。通过杀伤所需的时间长度(时间依赖性)或提高浓度的效果(浓度依赖性)来确定杀伤率。持续效应包括抗生素后效应(post-antibiotic effect,pae)。pae是抗生素暴露之后对细菌生长的持续抑制。
[0355]
使用这些参数,可将抗生素分为3类:
[0356]
[0357][0358]
对于i类抗生素(氨基糖苷类(ag's)、氟喹诺酮类、达托霉素和酮内酯类),理想的给药方案是使浓度最大化,因为浓度越高,杀伤的程度就越广泛且越快速。因此,24小时-auc/mic比和峰值/mic比是抗生素效力的重要预测指标。对于氨基糖苷类,最好是具有至少8-10的峰值/mic比以避免抗性。对于氟喹诺酮类与革兰阴性细菌,最佳的24小时-auc/mic比是约125。相比革兰阳性,40似乎是最佳的。然而,对于氟喹诺酮类,理想的24小时-auc/mic比在文献中差异很大。
[0359]
ii类抗生素(β-内酰胺类、克林霉素、红霉素、碳青霉烯类和利奈唑胺)显示出完全相反的特性。对于这些抗生素的理想给药方案是使暴露的持续时间最大化。t>mic是与效力最相关的参数。对于内酰胺类和红霉素,当高于mic的时间是给药间隔的至少70%时可见最大杀伤。
[0360]
iii类抗生素(万古霉素、四环素类、阿奇霉素和达福普汀-奎奴普丁组合)具有混合的性质,它们具有时间依赖性杀伤和中等的持续效应。对于这些抗生素,理想的给药方案是使接受的药量最大化。因此,24小时-auc/mic比是与效力相关联的参数。对于万古霉素,必需要有至少125的24小时-auc/mic比。
[0361]
患者,包括但不限于:患有由美罗培南敏感的细菌引起的呼吸道感染的cf、c0pd、非cf支气管扩张、吸入性肺炎、哮喘和vap患者,可以从这样的治疗中获益。碳青霉烯类抗生素的实例是:亚胺培南、培尼培南(panipenam)、美洛培南、多尼培南(doripenem)、比阿培南(biapenam)、mk-826、da-1131、er-35786、来那培南(lenapenam)、s-4661、cs-834(r-95867的前药)、kr-21056(kr-21012的前药)、l-084(ljc 11036的前药)和cxa-101。可通过先施用或共施用本发明的化合物和方法来增强所描述的所有抗感染剂的治疗效力。
[0362]
粘液溶解剂与示例性抗炎剂的组合:
[0363]
吸入式皮质类固醇是哮喘、copd以及其它呼吸系统疾病之慢性护理的标准,所述疾病的特征在于导致气流受限的急性和慢性炎症。适于与本发明中所述的方法和分子组合施用的抗炎剂的实例包括倍氯米松、布地奈德和氟替卡松以及一组不含类固醇的抗炎药,称为非甾体抗炎药(nsaid)。
[0364]
花生四烯酸代谢产物的产品尤其是白三烯类(lt),对肺部炎症有帮助。半胱氨酰白三烯(ltc 4、ltd 4和lte 4)主要由嗜酸性粒细胞、肥大细胞和巨噬细胞产生。适于通过本发明的方法施用的白三烯调节剂的实例包括孟鲁司特、齐留通和扎鲁司特。
[0365]
肥大细胞稳定剂是色甘酸类药物诸如色甘酸(色甘酸钠),其用于预防或控制某些过敏性病症。它们阻断肥大细胞脱粒所必需的钙通道、稳定细胞,从而防止组胺和相关介质
的释放。它们作为吸入剂用于治疗哮喘,作为鼻喷雾剂来治疗枯草热(过敏性鼻炎)以及作为滴眼剂用于过敏性结膜炎。最后,它们以口服形式用于治疗肥大细胞增多症的罕见病症。
[0366]
已显示pde4抑制剂调节肺部炎症并用于治疗慢性阻塞性肺病。适合于与本发明中所述的方法和分子组合使用的pde4抑制剂的实例包括但不限于茶碱和罗氟司特。
[0367]
粘液溶解剂与示例性支气管扩张剂的组合:
[0368]
一氧化氮(no)供体:no、no供体、no以及过氧亚硝酸盐清除剂和诱导型no合酶活性调节剂。一氧化氮是可通过吸入外源性施用的高效的内源性血管扩张剂和支气管扩张剂。其是通过血管内皮细胞钙依赖性酶一氧化氮合成酶由l-精氨酸的末端胍氮原子的转化合成,然后扩散穿过细胞膜以活化酶鸟苷酸环化酶。这种酶增强了环磷鸟苷(cgmp)的合成,引起血管和支气管平滑肌的松弛以及血管的舒张(palmer,circ res.,82(8):852-61(1998))。
[0369]
在内衬于血管的血管内皮细胞中合成的一氧化氮具有对于维持健康的呼吸和心血管系统来说重要的广泛功能(megson il等expert opin investig drugs.2002may;11(5):587-601.)。一氧化氮可用性的降低与许多疾病的发生和进展相关,并且递送补充的一氧化氮以帮助预防疾病进展是有吸引力的治疗选择。一氧化氮供体药物代表全身性一氧化氮递送的一种有用的方法并且多年来已使用有机硝酸酯作为缓解心绞痛症状的有效治疗。然而,硝酸酯具有局限性并且自从发现一氧化氮是重要的生物介质后,已经出现许多替代性一氧化氮供体种类。
[0370]
在呼吸道中,通过居留细胞和炎性细胞产生no(ricciardolo fl等,curr drug targets 2006jun;7(6):721-35)。通过由酶no合酶(nos)催化的l-精氨酸氧化而产生no。nos以三种不同的同种型存在:神经元型nos(nnos)、诱导型nos(inos)和内皮型nos(enos)。来源于nos的组成型同种型(nnos和enos)和其它no加合物分子(亚硝基硫醇(nitrosothiol))的no能够调节支气管肌紧张(bronchomotor tone)。通过nf-kb依赖性途径被不同的细胞因子上调的来源于no合酶的诱导型同种型的no似乎是具有免疫调节作用的促炎介质。在年老的cf患者中,inos的表达被显著降低(yoon等,j clin invest.2006feb;116(2):436-46)。慢性cf中inos的这种降低的表达与铜绿假单胞菌的类粘蛋白muc突变亚群的出现相关。已表明在cf气道中在ph6.5下,15mm no
2-杀伤muca铜绿假单胞菌。no本身或作为亚硝酰基铁种类的前体与这种抗微生物作用相关。因此,吸入的no
2-包括但不限于吸入的nano2,作为cf疗法具有吸引力。氧化应激条件下的no的产生间接地生成了可以放大哮喘和copd中的炎性应答的强氧化剂(活性氮(reactive nitrogen species))。此外,no可被呼出,在稳定的特应性哮喘以及在哮喘和copd二者加重期间no水平异常。因此,呼出的no可能是监测潜在的炎性过程的非侵入性工具。这表明nos调节提供了气道慢性炎性疾病诸如哮喘和copd的预防和治疗中的新型靶标。
[0371]
适合与本发明中所述的方法和分子组合施用的no、no供体和no合酶活性调节剂的实例包括在vallance等,fundam clin pharmacol.2003 feb;17(1):1-10,al-sa'doni hh等.mini rev med chem.2005 mar;5(3):247-54,miller mr等.br j pharmacol.2007 jun;151(3):305-21.epub 2007 apr 2以及katsumi h等cardiovasc hematol agents med chem.2007 jul;5(3):204-8中公开的吸入型no药剂。
[0372]
在某些条件下,诱导型no合酶的活性导致过量产生no,这继而又增加了炎症和组
织损伤。在这些条件下,与本发明中所述的方法和分子组合施用的以下的诱导型no合酶抑制剂、no清除剂和过氧亚硝酸盐/酯清除剂是合适的:bonnefous等,j.med.chem.,2009,52(9),第3047-3062页,muscara等ajp-gijune 1999第276卷,第6期g1313-g1316或hansel等faseb journal.2003;17:1298-1300。
[0373]
β2-肾上腺素能受体激动剂:已确定施用超治疗浓度的受体激动剂导致受体脱敏和效力损失。例如,对于基于β2-肾上腺素受体的支气管扩张剂已描述了这种现象(duringer等,br j pharmacol.,158(1):169-79(2009))。高浓度的这些受体激动剂导致受体磷酸化、内化和潜在的降解。在8至24小时的过程中或过夜经鼻插管通过吸入向患者施用受体激动剂,这造成通过快速喷雾器大剂量施用后的快速耐受,改善了此类药剂的效力,原因是降低了快速耐受的程度。β2肾上腺素能受体激动剂包括沙丁胺醇、左旋沙丁胺醇、沙丁胺醇、丙卡特罗、特布他林、吡布特罗和奥西那林。可通过先施用或共施用本发明的化合物和方法来增强β2-肾上腺素能受体激动剂的治疗效力。
[0374]
粘液溶解剂与示例性基因载体的组合:
[0375]
用于施用基因治疗的基因载体的实例包括病毒、dna:蛋白质复合物、质粒、dna和rna。
[0376]
粘液溶解剂与其它示例性治疗剂的组合:
[0377]
适合与本发明中所述的方法和分子组合施用的其它类的治疗剂的实例包括抗病毒剂,诸如利巴韦林;抗真菌剂,诸如两性霉素、伊曲康唑和伏立康唑;免疫抑制剂;抗排斥药物,诸如环孢素、他克莫司和西罗莫司;支气管扩张剂,包括但不限于抗胆碱能剂,诸如异丙托铵、噻托溴胺、阿地溴铵(aclidinium)等;pde5抑制剂sirna;基因治疗载体;适体;内皮素受体拮抗剂;α-1-抗胰蛋白酶;前列环素;疫苗;pde-4和pde-5抑制剂;以及类固醇,诸如倍氯米松、布地奈德、环索奈德、氟尼缩松、氟替卡松、莫米松和曲安西龙。
[0378]
实验方法和生物学测定:
[0379]
材料和方法
[0380]
除非另有说明,否则所有商业材料均按原样使用。所有溶剂均为试剂级或hplc级。无水thf、meoh、ch2cl2购自sigma-aldrich,不经进一步干燥即可使用。所有反应在预纯化的干燥ar(g)的气氛下进行。在bruker avance-400仪上记录nmr光谱,除非另有说明,溶剂cdcl3、cd3od和dmso-d6均购自aldrich或cambridge isotope laboratories。以下缩写用于解释多重性:s=单峰,d=双峰,t=三重峰,q=四重峰,m=多重峰,br=宽。化学位移以相对于四甲基硅烷(tms)为内标的ppm报道。在biotage微波反应器上进行微波反应。除非另有说明,所有反应均在氩气氛下在烘箱干燥的玻璃器皿中进行。通过使用uv光(作为显色剂)和茚三酮溶液并加热(作为显影剂),通过在0.25mm e.merck硅胶板(60f-254)上进行的tlc监测反应。对于极性化合物,通过hplc和lcms分析监测反应。使用e.merck硅胶(60,粒径0.040-0.063mm)进行快速柱色谱法。
[0381]
lcms和hplc法:
[0382]
使用在shimadzu lcms-lc-20ad上于254nm检测的sunfire c18,2.1x50 mm分析柱(除非另外指定)获得lcms分析。使用以下时间程序,流速为每分钟0.40ml。
[0383]
使用在shimadzu hplc系统上于220nm检测的xterra ms c18柱5μ4.6
×
150mm分析柱(除非另外指定)获得hplc分析。
[0384]
1.s-3-(2-(双((2s,3r,4r,5r)-2,3,4,5,6-五羟基己基)氨基)乙氧基)苄基呋喃-2-硫代羧酸酯盐酸盐(9)的制备;
[0385]
方案1
[0386][0387]
3-(2-((叔丁氧基羰基)氨基)乙氧基)苯甲酸甲酯(3)的制备;向化合物1(10.5g,69.0mmol)在dmf(40ml)中的溶液中加入k2co3(19.0g,138mmol),并在室温下搅拌5分钟。向上述反应混合物中加入化合物2(24.1g,104mmol),并将反应混合物在室温下搅拌48小时。将反应混合物用水(300ml)稀释,用etoac(3
×
300ml)萃取。将合并的有机层浓缩,残余物通过柱色谱纯化,得到白色胶状的化合物3(18.0g,89%):1h nmr(400mhz,cdcl3)δ7.62(dt,j=8.0,1.3hz,1h),7.56-7.52(m,1h),7.34(t,j=7.9hz,1h),7.10-7.06(m,1h),5.04(brs,1h),4.06(t,j=5.2hz,2h),3.90(s,3h),3.58-3.50(m,2h),1.45(s,9h);esi ms m/z 296[m+h]
+
。
[0388]
(2-(3-(羟甲基)苯氧基)乙基)氨基甲酸叔丁酯(4)的制备;在0℃下,在15分钟内以小批量向化合物3(18.0g,61.0mmol)在thf(500ml)中的溶液中加入氢化锂铝(3.50g,91.5mmol)。将所得反应混合物在0℃下搅拌1小时,并在0℃用冰冷的水淬灭。将反应混合物用etoac(300ml)稀释并通过硅藻土垫过滤,并用etoac(2
×
300ml)洗涤硅藻土垫。将滤液真空浓缩,通过柱色谱纯化,得到胶状固体的化合物4(14.0g,86%):1h nmr(400mhz,cd3cl)δ7.26(t,j=6.9hz,1h),6.96-6.90(m,2h),6.80(dd,j=8.5,2.5hz,1h),5.10(brs,1h),4.65(s,2h),4.01(t,j=5.1hz,2h),3.55-3.45(m,2h),1.97(brs,1h),1.44(s,9h);esi ms m/z 268[m+h]
+
。
[0389]
s-3-(2-((叔丁氧基羰基)氨基)乙氧基)苄基硫代乙酸酯(5)的制备;在0℃下向4(14.0g,52.4mmol)在ch2cl2(500ml)中的溶液中加入et3n(21.5ml,157mmol),然后加入甲磺酰氯(6.00ml,78.6mmol),并在室温搅拌1小时。将反应混合物用水(100ml)稀释,并用ch2cl2(3
×
150ml)萃取。将合并的有机萃取物用盐水洗涤,在na2so4上干燥,浓缩,得到黄色油状的甲磺酸化产物(20.0g,粗品),将其不经进一步纯化直接用于下一步骤:esi ms m/z 346[m+h]
+
。
[0390]
向dmf(50ml)和thf(250ml)中的粗甲磺酸化物(20.0g)中加入ksac(9.00g,78.8mmol)并在室温下搅拌16小时。除去溶剂,将残余物在水(100ml)与etoac(100ml)之间分配。分离出etoac层,水层用etoac(2
×
100ml)萃取。将合并的有机层浓缩,残余物通过柱色谱纯化,得到为黄色固体的化合物5(22.0g,两步62%):1h nmr(400mhz,cd3cl)δ7.19(t,j=7.7hz,1h),6.87(dd,j=7.6,1.5hz,1h),6.83-6.80(m,1h),6.76(dd,j=6.8,2.7hz,1h),4.99(brs,1h),4.08(s,2h),3.99(t,j=5.1hz,2h),3.56-3.44(m,2h),2.34(s,3h),1.45(s,9h);esi ms m/z 326[m+h]
+
。
[0391]
叔丁基(2-(3-(巯基甲基)苯氧基)乙基)氨基甲酸酯(6)的制备;
[0392]
向5(4.00g,12.7mmol)在thf(20ml)、甲醇(20ml)和水(20ml)的混合物中的溶液中加入固体lioh
·
h2o(1.06g,25.4mmol),并将反应混合物在室温下搅拌1小时。向上述反应混合物中加入tcep
·
hcl(1.81g,6.34mmol)并再搅拌1小时。除去溶剂,将残余物溶于etoac(50ml)中,并将溶液用饱和nahco3水溶液(40ml)洗涤。分离出etoac层,水层用etoac(2
×
150ml)萃取。将合并的有机层经na2so4干燥,过滤并浓缩,得到粗硫醇6(3.40g,95%,黄色液体),无需进一步纯化直接用于下一步骤。1h nmr(400mhz,cd3cl)δ7.22(t,j=8.3hz,1h),6.91(dd,j=8.1,1.1hz,1h),6.88-6.85(m,1h),6.76(dd,j=8.5,2.3hz,1h),4.98(brs,1h),4.01(t,j=5.5hz,2h),3.70(t,j=7.6hz,2h),3.56-3.48(m,2h),1.76(t,j=7.6hz,1h),1.45(s,9h);
[0393]
esi ms m/z 284[m+h]
+
。
[0394]
s-3-(2-((叔丁氧基羰基)氨基)乙氧基)苄基呋喃-2-硫代羧酸酯(7)的制备;在0℃下向化合物6(3.40g,120mmol)和et3n(5.05ml,36.0mmol)在ch2cl2(60ml)中的溶液中逐滴加入2-糠酰氯(1.78ml,18.0mmol),并在室温下搅拌1小时。向反应混合物中加入水(20ml),用ch2cl2(3
×
60ml)萃取。将合并的有机萃取物用盐水洗涤,经na2so4干燥,过滤,浓缩,残余物通过柱色谱纯化,得到为无色液体的化合物7(4.40g,97%):1h nmr(400mhz,cd3cl)δ7.58-7.54(m,1h),7.24-7.17(m,2h),6.95(dd,j=7.6,1.1hz,1h),6.92-6.88(m,1h),6.77(dd,j=8.2,2.3hz,1h),6.52(dd,j=3.5,1.5hz,1h),4.99(brs,1h),4.25(s,2h),4.00(t,j=4.4hz,2h),3.56-3.37(m,2h),1.44(s,9h);esi ms m/z 378[m+h]
+
。
[0395]
s-3-(2-氨基乙氧基)苄基呋喃-2-硫代羧酸酯盐酸盐(8)的制备;将化合物7(4.40g,11.7mmol)在室温下溶于二噁烷(50ml)中的4n hcl,将溶液搅拌1小时。浓缩后,将残余物用etoac研磨,过滤分离,得到为灰白色固体的盐酸盐8(3.40g,93%):1h nmr(400mhz,cd3od)δ7.77-7.74(m,1h),7.26(dd,j=3.9,1.2hz,1h),7.24(d,j=8.0hz,1h),7.04-7.02(m,1h),7.00(dd,j=7.8,1.1hz,1h),6.89(ddd,j=8.3,2.7,1.1hz,1h),6.64(dd,j=3.7,1.1hz,1h),4.27(s,2h),4.21(dd,j=4.8,2.1hz,2h),3.34(dd,j=4.8,1.2hz,2h);esi ms m/z 278[m+h]
+
。
[0396]
s-3-(2-(双(2s,3r,4r,5r)-2,3,4,5,6-五羟基己基)氨基)乙氧基)苄基呋喃-2-硫代羧酸酯盐酸盐{(9)}的制备;向胺8(1.00g,3.20mmol)在甲醇(50ml)中的溶液中加入d-葡萄糖(2.30g,12.7mmol)和乙酸(0.76ml,12.7mmol),然后加入氰基硼氢化钠(800mg,12.7mmol),将所得反应混合物加热至50℃,并在50℃下搅拌4小时。另外,加入d-葡萄糖(0.58g,3.20mmol)、乙酸(0.19ml,3.20mmol)和氰基硼氢化钠(200mg,3.20mmol),将所得反应混合物在50℃再搅拌1小时。在减压下除去溶剂,使用c18 gold柱将残余物通过反相色谱
法纯化。使用于水中的4n hcl将产物级分的ph调节至ph=3,然后合并,通过冻干除去溶剂,得到为灰白色固体的hcl盐9(1.20g,59%):1h nmr(400mhz,cd3od)δ7.76-7.74(m,1h),7.27(dd,j=3.4,1.4hz,1h),7.24(t,j=7.8hz,1h),7.06(d,j=1.2hz,1h),7.01(d,j=7.4hz,1h),6.93(dd,j=8.5,2.8hz,1h),6.64(dd,j=3.8,1.9hz,1h),4.44-4.38(m,2h),4.27(s,2h),4.26-4.18(m,2h),3.97-3.50(m,16h);1h nmr(400mhz,dmso-d6)δ8.85-8.68(m,1h),8.03(d,j=1.0hz,1h),7.41(d,j=3.7hz,1h),7.26(t,j=7.8hz,1h),7.00(d,j=8.0,1.5hz,2h),6.91(dd,j=7.9,1.8hz,1h),6.76(dd,j=3.8,1.6hz,1h),5.21-4.50(m,11h),4.44-4.31(m,2h),4.28(s,2h),4.23-4.16(m,1h),4.13-3.99(m,2h),3.84-3.22(m,16h);esi ms m/z 606[c
26
h
39
no
13
s+h]
+
。
[0397]
2.化合物s-3-(2-(双((2r,3r,4r,5r)-2,3,4,5,6-五羟基己基)氨基)乙氧基)苄基呋喃-2-硫代羧酸酯盐酸盐(10)的制备;
[0398]
方案2
[0399][0400]
向胺8(1.00g,3.20mmol)在甲醇(50ml)中的溶液中加入d-甘露糖(2.30g,12.7mmol)和乙酸(0.76ml,12.7mmol),随后加入氰基硼氢化钠(800mg,12.7mmol),将所得混合物在50℃下加热并搅拌4小时。另外,加入d-甘露糖(0.58g,3.20mmol)、乙酸(0.19ml,3.20mmol)和氰基硼氢化钠(200mg,3.20mmol),所得反应混合物在50℃再加热1小时。此时,另外加入d-甘露糖(0.58g,3.20mmol)、乙酸(0.19ml,3.20mmol)和氰基硼氢化钠(200mg,3.20mmol),将所得混合物在50℃下加热1小时。将反应混合物冷却至室温,加入水(20ml);在减压下除去溶剂后,加入额外的水(20ml),然后将固体沉淀从溶液中落下,并通过滤纸过滤,用水/甲醇洗涤得到10的游离碱。然后将游离碱用在水中的4n hcl酸化以制备hcl盐并冻干,得到为灰白色固体的10(1.38g,67)。1h nmr(400mhz,cd3od)δ7.77-7.74(m,1h),7.27(dd,j=3.8,0.8hz,1h),7.24(t,j=8.2hz,1h),7.06(d,j=1.8hz,1h),7.01(d,j=7.6hz,1h),6.94(dd,j=8.2,2.4hz,1h),6.64(dd,j=3.9,1.7hz,1h),4.46-4.36(m,2h),4.27(s,2h),4.19-4.08(m,2h),3.94-3.59(m,14h),3.55-3.43(m,2h);1h nmr(400mhz,dmso-d6)δ8.87-8.68(m,1h),8.05-8.01(m,1h),7.41(d,j=3.6hz,1h),7.26(t,j=7.8hz,1h),7.05-7.00(m,1h),6.99(d,j=8.2hz,1h),6.96-6.89(m,1h),6.75(dd,j=3.7,1.9h,1h),5.40-4.52(m,8h),4.46-4.33(m,2h),4.28(s,2h),4.22-3.82(m,5h),3.97-3.16(m,15h);esi ms m/z 606[c
26
h
39
no
13
s+h].
+
[0401]
3.s-3-(2-(双((2s,3r,4r,5r)-2,3,4,5,6-五羟基己基)氨基)乙氧基)苄基2-甲基硫代丙酸酯盐酸盐(14)的制备;
[0402]
方案3
[0403][0404]
s-3-(2-((叔丁氧基羰基)氨基)乙氧基)苄基2-甲基硫代丙酸酯(12)的制备;
[0405]
在0℃向化合物6(2.60g,9.18mmol)和et3n(3.75ml,27.5mmol)在ch2cl2(50ml)中的溶液中滴加异丁酰氯(11,1.45ml,13.8mmol)并在室温下搅拌1小时。向反应混合物中加入水(20ml),用ch2cl2(3
×
50ml)萃取。将合并的有机萃取物用盐水洗涤,经na2so4干燥,过滤,浓缩,残余物通过柱色谱纯化,得到为灰白色固体的化合物12(3.00g,93%):1h nmr(400mhz,cd3cl)δ7.19(t,j=7.7hz,1h),6.88(dd,j=7.6,1.6hz,1h),6.82(t,j=2.3hz,1h),6.76(dd,j=8.2,2.6hz,1h),4.98(brs,1h),4.06(s,2h),3.99(t,j=5.3hz,2h),3.55-3.45(m,2h),2.26(sep,j=7.1hz,1h),1.45(s,9h),1.20(d,j=7.1hz,6h);esi ms m/z 354[m+h]
+
。
[0406]
s-3-(2-氨基乙氧基)苄基2-甲基硫代丙酸酯盐酸盐(13)的制备;
[0407]
将化合物12(3.00g,8.40mmol)在室温下溶于二噁烷(30ml)中的4n hcl,搅拌该溶液1小时。浓缩后,残余物用etoac研磨,得到为灰白色固体状的盐酸盐13(2.25g,93%):1h nmr(400mhz,cd3od)δ7.23(t,j=7.5hz,1h),6.97-6.94(m,1h),6.88(dd,j=7.6,1.6hz,1h),6.92(dd,j=7.9,2.1hz,1h),4.21(t,j=5.1hz,2h),4.08(s,2h),3.35(t,j=5.4hz,2h),2.75(sep,j=7.1hz,1h),1.16(d,j=7.1hz,6h);esi ms m/z 254[m+h]
+
。
[0408]
s-3-(2-(双((2s,3r,4r,5r)-2,3,4,5,6-五羟基己基)氨基)乙氧基)苄基2-甲基硫代丙酸酯盐酸盐(14)的制备;
[0409]
向胺13(935mg,3.20mmol)在甲醇(50ml)中的溶液中加入d-葡萄糖(2.30g,12.7mmol)和乙酸(0.76ml,12.7mmol),然后加入氰基硼氢化钠(800mg,12.7mmol),将所得反应混合物在50℃下加热并搅拌4小时。另外,加入d-葡萄糖(0.58g,3.20mmol)和乙酸(0.19ml,3.20mmol)和氰基硼氢化钠(200mg,3.20mmol),将所得反应混合物在50℃下加热搅拌1小时。在减压下除去溶剂,使用c18gold柱将残余物通过反相色谱法纯化。使用于水中的4n hcl将产物级分的ph调节至ph=3,然后合并,通过冻干除去溶剂,得到为灰白色固体的14(1.50g,76%):1h nmr(400mhz,cd3od)δ7.23(t,j=8.3hz,1h),6.97(t,j=1.9hz,1h),6.92(td,j=7.4,2.4hz,2h),4.44-4.36(m,2h),4.29-4.18(m,2h),4.08(s,2h),3.93-3.50(m,16h),2.75(sep,j=7.2hz,1h),1.17(d,j=7.2hz,6h);1h nmr(400mhz,dmso-d6)δ8.72
(brs,1h),7.24(t,j=7.5hz,1h),6.96-6.87(m,3h),5.42-4.89(m,10h),4.42-4.31(m,2h),4.24-4.14(m,1h),4.12-4.03(m,2h),4.08(s,2h),3.78-3.64(m,4h),3.62-3.55(m,2h),3.55-3.27(m,10h),2.77(sep,j=7.0hz,1h),1.12(d,j=7.0hz,6h);esi ms m/z 582[c
25
h
43
no
12
s+h]
+
。
[0410]
4.s-3-(2-(双((2r,3r,4r,5r)-2,3,4,5,6-五羟基己基)氨基)乙氧基)苄基2-甲基硫代丙酸酯盐酸盐(15)的制备;
[0411]
方案4
[0412][0413]
向胺13(935mg,3.20mmol)在甲醇(50ml)中的溶液中加入d-甘露糖(2.30g,12.7mmol)和乙酸(0.76ml,12.7mmol),然后加入氰基硼氢化钠(800mg,12.7mmol),将所得反应混合物在50℃下加热并搅拌4小时。另外,加入d-甘露糖(0.58g,3.20mmol)、乙酸(0.19ml,3.20mmol)和氰基硼氢化钠(200mg,3.20mmol),将所得反应混合物在50℃再加热1小时。1小时后,加入另外的d-甘露糖(0.58g,3.20mmol)、乙酸(0.19ml,3.20mmol)和氰基硼氢化钠(200mg,3.20mmol),将所得混合物加在50℃下加热1小时。将反应混合物冷却至室温,加入水(20ml);在减压下除去溶剂,然后加入另外的水(20ml)。将沉淀的固体过滤并用水/甲醇洗涤以得到游离碱15的硼复合物。然后将游离碱用在水中的4n hcl酸化,制备盐然后冷冻干燥,得到为灰白色固体的15a(1.25g,63%)。1h nmr(400mhz,cd3od)δ7.22(t,j=7.5hz,1h),6.99-6.96(m,1h),6.95-6.90(m,2h),4.43-4.37(m,2h),4.20-4.09(m,2h),4.08(s,2h),3.93-3.61(m,14h),3.55-3.44(m,2h),2.75(sep,j=7.2hz,1h),1.17(d,j=7.2hz,6h);1h nmr(400mhz,dmso-d6)δ8.52(brs,1h),7.24(t,j=8.4hz,1h),6.98-6.88(m,3h),5.29-4.54(m,10h),4.42-4.33(m,2h),4.24-3.91(m,5h),4.08(s,2h),3.81-3.21(m,14h),2.77(sep,j=7.0hz,1h),1.12(d,j=7.0hz,6h);esi ms m/z 582[c
25
h
43
no
12
s+h]
+
。
[0414]
5.s-3-(2-(双((2s,3r,4r,5r)-2,3,4,5,6-五羟基己基)氨基)乙氧基)苄基硫代乙酸酯盐酸盐(17)的制备;
[0415]
方案5
[0416][0417]
s-3-(2-氨基乙氧基)苄基硫代乙酸酯盐酸盐(16)的制备;将化合物5(4.00g,粗品,12.7mmol)在室温下溶于二噁烷(40ml)中的4n hcl,将该溶液搅拌1小时。浓缩后,将残
余物用etoac研磨,然后通过过滤分离,得到为灰白色固体的盐酸盐16(2.60g,79%):1h nmr(400mhz,cd3od)δ7.23(t,j=7.9hz,1h),6.96-6.94(m,1h),6.93(dd,j=7.9,1.6hz,1h),6.88(ddd,j=8.4,2.5,1.1hz,1h),4.20(t,j=5.0hz,2h),4.09(s,2h),3.35(t,j=5.0hz,2h),2.32(s,3h);esi ms m/z 226[m+h]
+
。
[0418]
s-3-(2-(双((2s,3r,4r,5r)-2,3,4,5,6-五羟基己基)氨基)乙氧基)苄基硫代乙酸酯盐酸盐(17)的制备;
[0419]
向胺16(835mg,3.20mmol)在甲醇(50ml)中的溶液中加入d-葡萄糖(2.30g,12.7mmol)和乙酸(0.76ml,12.7mmol),然后加入氰基硼氢化钠(800mg,12.7mmol),将所得反应混合物在50℃加热4小时。另外加入d-葡萄糖(0.58g,3.20mmol)、乙酸(0.19ml,3.20mmol)和氰基硼氢化钠(200mg,3.20mmol),将所得反应混合物在50℃下加热1小时。在减压下除去溶剂,使用c18 gold柱将残余物通过反相色谱法纯化。用于水中的4n hcl将产物级分的ph调节至ph=3,然后合并,通过冻干除去溶剂,得到为白色固体的17(1.10g,58%)。1h nmr(400mhz,cd3od)δ7.24(t,j=7.8hz,1h),6.99-6.96(m,1h),6.95-6.89(m,2h),4.42-4.37(m,2h),4.28-4.18(m,2h),4.09(s,2h),3.92-3.50(m,16h),2.32(s,3h);1h nmr(400mhz,dmso-d6)δ8.66(brs,1h),7.24(t,j=8.5hz,1h),6.96-6.87(m,3h),5.89-5.16(m,2h),4.63-4.15(m,10h),4.13-4.01(m,3h),4.09(s,2h),3.78-3.66(m,4h),3.63-3.55(m,2h),3.54-3.26(m,10h),2.35(s,3h);esi ms m/z 554[c
23
h
39
no
12
s+h]
+
。
[0420]
6.s-3-(2-(双((2r,3r,4r,5r)-2,3,4,5,6-五羟基己基)氨基)乙氧基)苄基硫代乙酸酯(18)的制备;
[0421]
方案6
[0422][0423]
化合物18的制备;
[0424]
向胺16(835mg,3.20mmol)在甲醇(50ml)中的溶液中加入d-葡萄糖(2.30g,12.7mmol)和乙酸(0.76ml,12.7mmol),然后加入氰基硼氢化钠(800mg,12.7mmol),将所得混合物在50℃下加热4小时。另外加入d-葡萄糖(0.58g,3.20mmol)、乙酸(0.19ml,3.20mmol)和氰基硼氢化钠(200mg,3.20mmol),将所得混合物在50℃下加热1小时。加入第二个当量的d-甘露糖(0.58g,3.20mmol)、乙酸(0.19ml,3.20mmol)和氰基硼氢化钠(200mg,3.20mmol),将所得混合物在50℃加热1小时。将反应混合物冷却至室温,加入水;在减压下除去溶剂后,加入更多的水并沉淀出固体,并用水/甲醇过滤洗涤,得到游离碱18的硼复合物。然后将游离碱复合物用于水中的4n hcl酸化以制备hcl盐并冻干得到为灰白色固体的18(通过hplc,纯度为90%)。使用c18 gold柱,通过反相色谱法进一步纯化固体,得到为白色固体的18(450mg,24%),并通过hplc纯化,分离出另外1.00g 18,纯度<95%。1h nmr(400mhz,cd3od)δ7.22(t,j=7.8hz,1h),6.99-6.97(m,1h),6.95-6.90(m,2h),4.42-4.37
(m,2h),4.19-4.11(m,2h),4.09(s,2h),3.94-3.61(m,14h),3.55-3.44(m,2h),2.32(s,3h);1h nmr(400mhz,dmso-d6)δ8.42(brs,1h),7.25(t,j=7.9hz,1h),6.93-6.86(m,3h),5.71-5.57(m,1h),5.54-5.42(m,1h),4.92-4.15(m,11h),4.09(s,2h),4.05-3.87(m,3h),3.77-3.21(m,14h),2.35(s,3h);esi ms m/z 554[c
23
h
39
no
12
s+h].
+
[0425]
7.(2r,2'r,3r,3'r,4r,4'r,5s,5's)-6,6'-((2-(3-(巯基甲基)苯氧基)乙基)氮烷二基)双(己烷-1,2,3,4,5-五醇)盐酸盐(19)的制备;
[0426]
方案7
[0427][0428]
向9(350mg,0.54mmol)在水(15ml)中的溶液中加入固体lioh
·
h2o(69mg,1.63mmol),将反应混合物在室温下搅拌1小时。向该混合物中加入tcep
·
hcl(154mg,0.54mmol)并搅拌1小时。通过4n hcl水溶液使反应的ph值达到ph=2,除去溶剂。类似地,用lioh
·
h2o、tcep
·
hcl处理335mg(0.54mmol)14和150mg(0.25mmol)17,用4n hcl酸化后,与来自9的产物合并,并通过反相柱色谱纯化,然后冻干至得到550mg(75%)为吸湿性白色固体的纯化合物19:1h nmr(400mhz,cd3od)δ7.23(t,j=7.8hz,1h),7.05-7.01(m,1h),6.98(d,j=8.1hz,1h),6.90(dd,j=8.1,2.4hz,1h),4.40-4.37(m,2h),4.30-4.17(m,2h),3.96-3.48(m,18h);1h nmr(400mhz,dmso-d6)δ8.67(brs,1h),7.24(t,j=7.8hz,1h),7.01-6.98(m,1h),6.96(d,j=8.0hz,1h),6.87(dd,j=8.0,2.5hz,1h),4.72-4.15(m,10h),4.14-4.02(m,2h),3.78-3.66(m,7h),3.64-3.55(m,2h),3.53-3.28(m,11h),2.88(t,j=7.7hz,1h);esi ms m/z 512[c
21
h
37
no
11
s+h]
+
。
[0429]
8.(2r,2'r,3r,3'r,4r,4'r,5r,5'r)-6,6'-((2-(3-(巯基甲基)苯氧基)乙基)氮烷二基)双(己烷-1,2,3,4,5-五醇)盐酸盐(20)的制备;
[0430]
方案8
[0431][0432]
向10(350mg,0.54mmol)在水(15ml)中的溶液中加入固体lioh
·
h2o(69mg,
1.63mmol),将混合物在室温下搅拌1小时。向反应混合物中加入tcep
·
hcl(30.0mg,0.10mmol)并搅拌1小时。通过4n hcl水溶液使上述反应混合物的ph达到ph=2,除去溶剂。类似地,用lioh
·
h2o,tcep
·
hcl处理220mg(0.35mmol)15和1.00g(1.70mmol)18,并在用4n hcl酸化后,与衍生自10的产物合并,并通过反相柱色谱纯化和冻干,得到750mg(53%)为吸湿性白色固体的纯化合物20:1h nmr(400mhz,cd3od)δ7.23(t,j=7.9hz,1h),7.04(t,j=1.9hz,1h),6.97(d,j=7.9hz,1h),6.91(dd,j=8.1,2.4hz,1h),4.46-4.37(m,2h),4.22-4.09(m,2h),3.96-3.61(m,16h),3.56-3.44(m,2h);1h nmr(400mhz,dmso-d6)δ8.54(brs,1h),7.24(t,j=7.9hz,1h),7.01(t,j=2.5hz,1h),6.96(d,j=7.9hz,1h),6.88(dd,j=7.9,2.5hz,1h),5.35-4.71(m,10h),4.47-4.28(m,2h),4.09-3.93(m,2h),3.78-3.23(m,17h),2.88(t,j=7.7hz,1h);esi ms m/z 512[c
21
h
37
no
11
s+h]
+
。
[0433]
9.s-3-(2-(双((2s,3r,4r,5r)-2,3,4,5,6-五羟基己基)氨基)乙氧基)苄基硫代丙酸酯盐酸盐(24)的制备;
[0434]
方案9
[0435][0436]
s-3-(2-((叔丁氧羰基)氨基)乙氧基)苄基硫代丙酸酯(22)的制备;在0℃向化合物6(2.70g,9.54mmol)和et3n(3.90ml,28.6mmol)在ch2cl2(50ml)中的溶液中滴加丙酰氯(21,1.29ml,14.3mmol),然后在室温下搅拌1小时。过滤固体,浓缩滤液。向反应混合物中加入水(20ml),用ch2cl2(3
×
50ml)萃取。将合并的有机萃取物用盐水洗涤,经na2so4干燥,浓缩,将残余物通过柱色谱纯化,得到为灰白色固体的化合物22(3.00g,两步93%):1h nmr(400mhz,cd3cl)δ7.19(t,j=8.0hz,1h),6.88(dd,j=7.7,1.3hz,1h),6.82(t,j=2.1hz,1h),6.76(dd,j=8.4,2.4hz,1h),4.98(brs,1h),4.08(s,2h),3.99(t,j=5.1hz,2h),3.56-3.465(m,2h),2.59(q,j=7.8hz,2h),1.45(s,9h),1.19(t,j=7.9hz,3h);esi ms m/z 340[m+h]
+
。
[0437]
s-3-(2-氨基乙氧基)苄基硫代丙酸酯盐酸盐(23)的制备;将化合物22(3.00g,8.84mmol)在室温下溶于二噁烷(30ml)中的4n hcl,搅拌该溶液1小时。浓缩后,将残余物用etoac研磨并通过过滤分离,得到为灰白色固体的盐酸盐23(2.20g,91%):1h nmr(400mhz,cd3od)δ7.22(t,j=8.0hz,1h),6.95(t,j=1.9hz,1h),6.92(d,j=7.6hz,1h),6.88(ddd,j=8.2,2.5,0.7hz,1h),4.21(t,j=5.1hz,2h),4.09(s,2h),3.35(t,j=5.1hz,2h),2.59
氨基丙基)喹喔啉-2-基)甲基)硫代乙酸酯盐酸盐(38)的制备;
[0445]
方案11
[0446][0447]
6-溴喹喔啉-2-羧酸乙酯(28)和7-溴喹喔啉-2-羧酸乙酯(29)的制备
[0448]
在氮气下,在室温下向搅拌的4-溴-邻苯二胺(10.0g,54.05mmol)的1-甲基-2-吡咯烷酮(50ml)溶液中滴加溴丙酮酸乙酯27(20.9g,108.1mmol)。20小时后,将反应混合物在水(100ml)和乙酸乙酯(100ml)之间分配。分离有机相,水相用乙酸乙酯(2
×
100ml)萃取。合并的有机萃取物用水(2
×
50ml)洗涤,经硫酸镁干燥,过滤,将滤液浓缩。粗产物通过硅胶快速色谱法纯化,得到区域异构体28和29的60:40%混合物(6.50g,43%),其通过hplc和lc-ms分析表征,并直接用于下一步骤。esi-lcms m/z 282(m+h)
+
。
[0449]
6-(3-((叔丁氧基羰基)氨基)丙基)喹喔啉-2-羧酸乙酯(31)和7-(3-((叔丁氧基羰基)氨基)丙基)喹喔啉-2-羧酸乙酯(32)的制备;在氩气下向化合物30(3.64g,23.21mmol)的无水thf(300ml)溶液中加入9-bbn(0.5m,在thf中,116ml,58.05mmol)。将反应混合物在室温下搅拌2小时后,在室温下加入化合物28和29(6.50g,23.2mmol)、pd(pph3)2cl2(814mg,1.16mmol)和2n na2co3水溶液(15ml)。将所得混合物搅拌1小时。在溶剂除去后;将残余物在etoac(200ml)和水(200ml)之间分配。分离水层,用etoac(2
×
200ml)萃取。合并的有机萃取物用盐水洗涤,经na2so4干燥并真空浓缩。将粗产物通过硅胶柱色谱纯化,然后用庚烷中的10%乙醇作为洗脱剂进行手性hplc(od),得到5.10g,61%(2.50g的32和1.75g的31),通过1h nmr和lc-ms分析对其进行表征。
[0450]
(31)1h nmr(cdcl3,300mhz):δ9.50(s,1h),8.22(d,j=8.8hz,1h),7.95(br s,1h),7.72-7.68(m,1h),4.62-4.55(m,3h),3.28-3.21(m,2h),2.93(t,j=7.4hz,2h),2.02-1.91(m,2h),1.50(t,j=7.2hz,3h),1.44(s,9h)。esi-lcms m/z 360(m+h)
+
。
[0451]
(32)1h nmr(cdcl3,300mhz):δ9.48(s,1h),8.05(d,j=8.8hz,2h),7.76-7.72(m,1h),4.62-4.53(m,3h),3.24-3.17(m,2h),2.92(t,j=7.6hz,2h),2.05-1.91(m,2h),1.50(t,j=7.2hz,3h),1.44(s,9h)。esi-lcms m/z 360(m+h)
+
。
[0452]
(3-(2-(羟基甲基)喹喔啉-6-基)丙基)氨基甲酸叔丁酯(33)的制备
[0453]
在0℃向化合物31(1.75g,4.87mmol)在etoh(100ml)中的溶液中加入硼氢化钠(926mg,24.3mmol)。将所得反应混合物在室温下搅拌3小时,并将残余物在etoac(100ml)和水(100ml)之间分配。分离水层,用etoac(2
×
100ml)萃取。将合并的有机萃取物用盐水洗涤,经na2so4干燥,过滤并真空浓缩。将粗产物通过柱色谱纯化,得到为棕色液体的化合物33(1.10g,71%),将其立即用于下一步骤;esi ms m/z 318[m+h].
+
[0454]
s-((6-(3-((叔丁氧羰基)氨基)丙基)喹喔啉-2-基)甲基)硫代乙酸酯(34)的制备
[0455]
在0℃下向33(1.10g,3.47mmol)在ch2cl2(100ml)中的溶液中加入et3n(0.6ml,4.16mmol),然后加入甲磺酰氯(490mg,4.16mmol),然后在室温下搅拌2小时。将反应混合物用水(100ml)稀释,用ch2cl2(3
×
150ml)萃取。将合并的有机萃取物用盐水洗涤,经na2so4干燥,浓缩,得到黄色油状的甲磺酸化产物(1.40g,粗品),其不经进一步纯化直接用于下一步骤:esi ms m/z 396[m+h]
+
。
[0456]
向粗制甲磺酸化物(1.40g,3.54mmol)在dmf(10ml)中的溶液中加入ksac(1.02g,8.85mmol),并在室温下搅拌2小时。除去溶剂,将残余物在水(100ml)和etoac(100ml)之间分配。分离出etoac层,水层用etoac(2
×
100ml)萃取。将合并的有机层浓缩,将残余物通过柱色谱纯化,得到为黄色固体的化合物34(700mg,两步54%):1h nmr(400mhz,cdcl3)δ8.84(s,1h),7.95(d,j=8.6hz,1h),7.86(br s,1h),7.61-7.59(m,1h),4.57br s,1h),4.44(s,2h),3.23-3.16(m,2h),2.88(t,j=7.4hz,2h),2.40(s,3h),1.96-1.89(m,2h),1.44(s,9h);esi ms m/z 376[m+h]
+
。
[0457]
s-((6-(3-氨基丙基)喹喔啉-2-基)甲基)硫代乙酸酯盐酸盐(35)的制备
[0458]
将化合物34(500mg,1.33mmol)溶于dcm(5ml)中,然后在室温下滴加于二噁烷(5ml)中的4n hcl,然后将溶液在室温下搅拌2小时。除去溶剂后,残余物用etoac/己烷研磨,然后通过过滤分离,得到为灰白色固体的盐酸盐35(300mg,83%):1h nmr(300mhz,cd3od)δ8.87(s,1h),8.01-7.91(m,2h),7.76-7.73(m,1h),4.46(s,2h),3.03-2.96(m,4h),
2.38(s,3h),2.14-2.07(m,2h);1h nmr(300mhz,dmso-d6)δ8.91(s,1h),7.99-7.91(m,5h),7.77-7.71(m,1h),4.47(s,2h),2.95-2.77(m,4h),2.39(s,3h),2.05-1.92(m,2h);esi ms m/z 276[m+h]
+
。
[0459]
(3-(3-羟基甲基)喹喔啉-6-基)丙基)氨基甲酸叔丁酯(36)的制备
[0460]
在0℃,向化合物32(1.15g,3.20mmol)在etoh(100ml)中的溶液中加入硼氢化钠(608mg,16.0mmol)。将所得反应混合物在室温下搅拌3小时,并将残余物在etoac(100ml)和水(100ml)之间分配。分离水层,用etoac(2
×
100ml)萃取。将合并的有机萃取物用盐水洗涤,经na2so4干燥并真空浓缩。将粗产物通过柱色谱纯化,得到为棕色液体的化合物36(800mg,79%),将其立即用于下一步骤;esi ms m/z 318[m+h]
+
。
[0461]
(7-(3-((叔丁氧基羰基)氨基)丙基)喹喔啉-2-基)甲基甲磺酸酯的制备;
[0462]
在0℃向36(800mg,2.52mmol)在ch2cl2(100ml)中的溶液中加入et3n(0.44ml,3.02mmol),然后加入甲磺酰氯(356mg,3.02mmol),然后在室温下搅拌2小时。将反应混合物用水(100ml)稀释,用ch2cl2(3
×
150ml)萃取。将合并的有机萃取物用盐水洗涤,经na2so4干燥,浓缩,得到黄色油状的甲磺酸化产物(680mg,68%),其不经进一步纯化直接用于下一步骤:esi ms m/z 396[m+h]
+
。
[0463]
s-((7-(3-((叔丁氧羰基)氨基)丙基)喹喔啉-2-基)甲基)硫代乙酸酯(37)的制备
[0464]
向dmf(5ml)中的粗产物36(680mg,1.72mmol,粗品)中加入ksac(490mg,4.30mmol),并在室温下搅拌2小时。除去溶剂,将残余物在水(100ml)和etoac(100ml)之间分配。分离出etoac层,水层用etoac(2
×
100ml)萃取。将合并的有机层浓缩,将残余物通过柱色谱纯化,得到为黄色固体的化合物37(420mg,66%);esi ms m/z 376[m+h]
+
。
[0465]
s-((7-(3-氨基丙基)喹喔啉-2-基)甲基)硫代乙酸酯盐酸盐(38)的制备
[0466]
将化合物37(150mg,0.4mmol)溶于dcm(5ml)中,然后在室温下逐滴加入于二噁烷(5ml)中的4n hcl,并将溶液在相同温度下搅拌2小时。除去溶剂后,将残余物用etoac/己烷洗涤,然后通过过滤分离,得到为灰白色固体的盐酸盐38(66mg,61%):1h nmr(300mhz,cd3od)δ8.85(s,1h),8.06-7.99(m,1h),7.88(br s,1h),7.74-7.71(m,1h),4.47(s,2h),3.03-2.96(m,4h),2.38(s,3h),2.15-2.07(m,2h);1h nmr(300mhz,dmso-d6)δ8.89(s,1h),8.03(d,j=8.4hz,1h),7.96-7.81(m,4h),7.73-7.77(m,1h),4.47(s,2h),2.92(t,j=7.8hz,2h),2.85-2.78(m,2h),2.38(s,3h),2.04-1.93(m,2h);esi ms m/z 276[m+h]
+
。
[0467]
12.s-4-((6-氨基己基)氧基)苄基硫代乙酸酯盐酸盐(45)和(2r,2'r,3r,3'r,4r,4'r,5s,5's)-6,6'-((6-(4-(巯基甲基)苯氧基)己基)氮烷二基(azanediyl)双(己烷-1,2,3,4,5-五醇)盐酸盐(47)的制备;
[0468]
方案12
[0469][0470]
4-((6-((叔丁氧基羰基)氨基)己基)氧基)苯甲酸乙酯(41)的制备;
[0471]
向化合物39(3.00g,18.0mmol)的dmf(80ml)溶液中加入cs2co3(11.7g,36.1mmol),并在室温下搅拌5分钟。向上述反应混合物中加入化合物40(10.1g,36.1mmol),将反应混合物在室温下搅拌20小时。将反应混合物用水(100ml)稀释,并用etoac(3
×
100ml)萃取。将合并的有机层浓缩,将残余物通过柱色谱纯化,得到为白色胶状物的化合物41(4.65g,70%),直接将其用于下一步骤;esi ms m/z 366[m+h]
+
。
[0472]
(6-(4-(羟基甲基)苯氧基)己基)氨基甲酸叔丁酯(42)的制备;在0℃下向化合物41(4.60g,12.6mmol)的thf(100ml)溶液中分批加入氢化铝锂(515mg,15.1mmol)。将所得反应混合物在0℃下搅拌1小时,并在0℃用冰冷的水淬灭。将反应混合物用etoac(200ml)稀释并通过硅藻土垫过滤,并将硅藻土垫用etoac(200ml)洗涤。将滤液真空浓缩,并通过柱色谱纯化,得到为胶状固体的化合物42(3.10g,76%),将其直接用于下一步;esi ms m/z 324[m+h]
+
。
[0473]
4-((6-((叔丁氧基羰基)氨基)己基)氧基)苄基甲磺酸酯(43)的制备;
[0474]
在0℃向42(3.10g,9.59mmol)在ch2cl2(100ml)中的溶液中加入et3n(1.66ml,11.5mmol),然后加入甲磺酰氯(1.35g,11.5mmol),然后在室温下搅拌2小时。将反应混合物用水(100ml)稀释,用ch2cl2(3
×
150ml)萃取。将合并的有机萃取物用盐水洗涤,经na2so4干燥,浓缩,得到黄色油状的甲磺酸化产物43(3.40g,89%),将其不经进一步纯化直接用于下一步骤:esi ms m/z 401[m+h]
+
。
[0475]
s-4-((6-((叔丁氧基羰基)氨基)己基)氧基)苄基硫代乙酸酯(44)的制备;
[0476]
向上述产物43(3.40g,8.47mmol)在dmf(100ml)中的溶液中加入ksac(2.41g,21.1mmol)并在室温下搅拌2小时。除去溶剂,将残余物在水(100ml)和etoac(100ml)之间分配。分离出etoac层,水层用etoac(2
×
100ml)萃取。将合并的有机层浓缩,将残余物通过柱色谱纯化,得到为黄色固体的化合物44(2.80g,87%):1h nmr(400mhz,cdcl3)δ7.18(d,j=8.6hz,2h),6.80(d,j=8.6hz,2h),4.49(br s,1h),4.07(s,2h),3.91(t,j=6.4hz,2h),3.15-3.07(m,2h),2.33(s,3h),1.77-1.72(m,2h),1.51-1.34(m,15h);esi ms m/z 381[m+h]
+
。
[0477]
s-4-((6-氨基己基)氧基)苄基硫代乙酸酯盐酸盐(45)的制备;
[0478]
将化合物44(2.80g,7.34mmol)在室温下溶于二噁烷(20ml)中的4n hcl,然后将该溶液搅拌1小时。除去溶剂后,将残余物用etoac研磨,然后过滤,得到为吸湿性灰白色固体的盐酸盐45(1.80g,87%):1h nmr(300mhz,cd3od)δ7.17(d,j=8.8,2h),6.80(d,j=8.8,2h),4.05(s,2h),3.96(t,j=8.8,6.2,2h),2.92(t,j=7.4,2h),2.30(s,3h),1.83-1.63(m,4h),1.59-1.43(m,4h),1h nmr 300mhz(dmso-d6):δ7.88(br s,3h),7.19(d,j=8.8,2h),6.84(d,j=8.8,2h),4.04(s,2h),3.93(t,j=6.4,2h),2.75(br s,2h),2.34(s,3h),1.73-1.51(m,4h),1.42-1.33(m,4h);esi ms m/z 282[m+h]
+
。
[0479]
s-4-((6-(双((2s,3r,4r,5r)-2,3,4,5,6-五羟基己基)氨基)己基)氧基)苄基硫代乙酸酯盐酸盐(46)的制备;
[0480]
向胺45(500mg,1.78mmol)在甲醇(50ml)中的溶液中加入d-葡萄糖(960mg,5.33mmol)和乙酸(0.32ml,5.33mmol),然后加入氰基硼氢化钠(335mg,5.33mmol),将所得混合物在室温下搅拌16小时。加入另外的d-葡萄糖(640mg,3.56mmol)、acoh(0.21ml,3.56mmol)和氰基硼氢化钠(223mg,3.56mmol),将混合物在室温下搅拌24小时。加入另外的d-葡萄糖(960mg,5.33mmol)、acoh(0.32ml,5.33mmol)和氰基硼氢化钠(335mg,5.33mmol),将混合物在室温下搅拌24小时。除去溶剂后,用nahco3溶液中和反应混合物,并通过反相色谱法纯化,得到为白色固体的纯46(710mg,66%)。
[0481]
(2r,2'r,3r,3'r,4r,4'r,5s,5's)-6,6'-((6-(4-(巯基甲基)苯氧基)己基)氮烷二基)双(己烷-1,2,3,4,5-五醇)盐酸盐(47)的制备;
[0482]
向化合物46(710mg,1.16mmol)的水(20ml)溶液中加入固体lioh
·
h2o(195mg,4.66mmol),将反应混合物在室温下搅拌1小时,之后加入固体tcep
·
hcl(165mg,0.58mmol),将反应混合物在室温下搅拌2小时。用4n hcl酸化反应物,将残余物通过反相柱色谱纯化,得到无色胶状固体的盐酸盐47(420mg,64%):1h nmr(300mhz,cd3od)δ7.22(d,j=8.6,2h),6.83(d,j=8.6,2h),4.16(br s,2h),3.98-3.96(m,2h),3.83-3.72(m,4h),3.71-3.59(m,8h),3.51-3.36(m,5h),1.90-1.72(m,4h),1.64-1.43(m,4h);1h nmr300mhz(dmso-d6):δ8.74(br s,1h),7.22(d,j=8.6,2h),6.84(d,j=8.6,2h),5.45-5.40(m,2h),
4.79-4.45(m,8h),4.00-3.91(m,4h),3.70-3.64(m,4h),3.62-3.56(m,2h),3.51-3.38(m,7h),3.26-3.14(m,5h),2.73(t,j=7.4,1h),1.77-1.64(m,4h),1.49-1.29(m,4h);esi ms m/z 568[m+h]
+
。
[0483]
13.化合物48和49的制备;
[0484]
方案13
[0485][0486]
化合物48和49的制备
[0487]
向45(200mg,0.71mmol)的水(50ml)溶液中加入固体lioh
·
h2o(120mg,2.84mmol),并在室温下搅拌1小时,然后加入tcep
·
hcl(406mg,1.42mmol),并反应混合物在室温下搅拌1小时。反应完成后,其用1n hcl酸化并通过反相色谱纯化,得到为灰白色固体的化合物48(80mg,47%)和49(50mg,25%)
[0488]
:1h nmr(300mhz,cd3od)δ7.22(d,j=8.6,2h),6.82(d,j=8.6,2h),3.96(t,j=6.2,2h),3.67(br s,2h),2.92(t,j=7.4,2h),1.84-1.63(m,4h),1.59-1.46(m,4h);1h nmr 300mhz(dmso-d6):δ7.85(br s,3h),7.22(d,j=8.6,2h),6.85(d,j=8.6,2h),3.93(t,j=6.6,2h),3.67(d,j=7.2,2h),2.78-2.71(m,3h),1.74-1.51(m,4h),1.46-1.34(m,4h)。esi ms m/z 240[m+h]
+
。
[0489]
(49):1h nmr(300mhz,cd3od)δ7.21(d,j=8.6,2h),6.82(d,j=8.6,2h),3.94(t,j=6.4,2h),3.67(br s,2h),3.16(t,j=6.6,2h),1.91(s,3h),1.78-1.71(m,2h),1.58-1.36(m,6h);1h nmr 300mhz(dmso-d6):δ7.75(br s,1h),7.21(d,j=8.6,2h),6.84(d,j=8.6,2h),3.92(d,j=6.6,2h),3.67(d,j=7.6,2h),3.04-2.97(m,3h),2.72(t,j=7.2,1h),1.77(s,3h),1.71-1.66(m,2h),1.41-1.34(m,6h).esi ms m/z 282[m+h]
+
。
[0490]
14.s-4-(2-(双((2s,3r,4r,5r)-2,3,4,5,6-五羟基己基)氨基)乙氧基)苄基2-甲基硫代丙酸酯盐酸盐(56);s-4-(2-(双((2r,3r,4r,5r)-2,3,4,5,6-五羟基己基)氨基)乙氧基)苄基2-甲基硫代丙酸酯盐酸盐(59);和(2r,2'r,3r,3'r,4r,4'r,5r,5'r)-6,6'-((2-(4-(巯基甲基)苯氧基)乙基)氮烷二基)双(己烷-1,2,3,4,5-五醇)盐酸盐(58)的制备;
[0491]
方案14
354[m+h]
+
。
[0497]
s-4-(2-氨基乙氧基)苄基2-甲基硫代丙酸酯盐酸盐(55)的制备;
[0498]
将化合物54(2.60g,7.36mmol)在室温下溶于二噁烷(26ml)中的4n hcl,并搅拌该溶液2小时。浓缩后,将残余物用etoac研磨,然后通过过滤分离,得到为灰白色固体的盐酸盐55(1.50g,81%);esi ms m/z 254[m+h]
+
。
[0499]
s-4-(2-(双((2s,3r,4r,5r)-2,3,4,5,6-五羟基己基)氨基)乙氧基)苄基2-甲基硫代丙酸酯盐酸盐(56)的制备;
[0500]
向胺55(1.70g,6.71mmol)在甲醇(75ml)中的溶液中加入d-葡萄糖(2.41g,13.4mmol)和乙酸(0.82ml,13.4mmol),然后加入氰基硼氢化钠(846mg,13.4mmol),将所得混合物在室温下在55℃下搅拌2小时。加入另外的d-葡萄糖(0.85g,3.36mmol)、acoh(0.41ml,3.36mmol)和氰基硼氢化钠(423mg,3.36mmol),将混合物在55℃下搅拌2小时。加入另外的d-葡萄糖(0.85g,3.36mmol)、acoh(0.41ml,3,36mmol)和氰基硼氢化钠(423mg,3.36mmol),将混合物在55℃下搅拌2小时。减压除去溶剂后,使用c18 gold柱将残余物通过反相色谱纯化,得到为白色固体的纯56(1.65g,42%)。1h nmr(400mhz,cd3od)δ7.23(d,j=8.6hz,2h),6.95(d,j=8.6hz,2h),4.42-4.35(m,2h),4.27-4.17(m,2h),4.05(s,2h),3.93-3.49(m,16h),2.77-2.70(m,1h),1.16(s,3h),1.15(s,3h);1h nmr(400mhz,dmso-d6)δ8.64(br s,1h),7.22(d,j=8.6hz,2h),6.94(d,j=8.6hz,2h),4.37-4.29(m,3h),4.26-3.82(m,12h),3.76-3.56(m,7h),3.54-3.27(m,10h),2.78-2.72(m,1h),1.12(s,3h),1.10(s,3h);esi ms m/z 582[m+h]
+
。
[0501]
s-4-(2-(双((2r,3r,4r,5r)-2,3,4,5,6-五羟基己基)氨基)乙氧基)苄基2-甲基硫代丙酸酯(57)的制备;
[0502]
向胺55(1.50g,5.92mmol)在甲醇(75ml)中的溶液中加入d-甘露糖(2.13g,1.8mmol)和乙酸(0.72ml,11.8mmol),然后加入氰基硼氢化钠(735mg,11.8mmol),并将所得混合物在室温下在55℃下搅拌2小时。加入另外的d-甘露糖(1.07g,5.93mmol)、acoh(0.36ml,5.93mmol)和氰基硼氢化钠(368mg,5.93mmol),将混合物在55℃下搅拌2小时。加入另外的d-甘露糖(1.07g,5.93mmol)、acoh(0.36ml,5.93mmol)和氰基硼氢化钠(368mg,5.93mmol),将混合物在55℃下搅拌2小时。除去溶剂后,将反应混合物从水:甲醇(100:20ml)中结晶,得到为白色固体的游离碱57的硼复合物(2.25g,66%)。esi ms m/z 582[m+h]
+
。
[0503]
s-4-(2-(双((2r,3r,4r,5r)-2,3,4,5,6-五羟基己基)氨基)乙氧基)苄基2-甲基硫代丙酸酯盐酸盐(59)的制备;
[0504]
在室温下将化合物57(500mg,0.89mmol)溶于4n当量的hcl(0.85ml)水溶液中,该溶液用水稀释并冻干,得到为吸湿性灰白色固体的盐酸盐59(490g,92%):1h nmr(400mhz,cd3od)δ7.23(d,j=8.6hz,2h),6.96(d,j=8.6hz,2h),4.42-4.36(m,2h),4.18-4.01(m,4h),3.93-3.58(m,14h),3.53-3.44(m,2h),2.77-2.70(m,1h),1.16(s,3h),1.15(s,3h).1h nmr(400mhz,dmso-d6)δ8.62(br s,1h),7.23(d,j=8.6hz,2h),6.95(d,j=8.6hz,2h),4.37(br s,4h),4.24-3.77(m,16h),3.76-3.15(m,18h),2.78-2.72(m,1h),1.12(s,3h),1.10(s,3h);esi ms m/z 582[m+h]
+
。
[0505]
(2r,2'r,3r,3'r,4r,4'r,5r,5'r)-6,6'-((2-(4-(巯基甲基)苯氧基)乙基)氮烷
二基)双(己烷-1,2,3,4,5-五醇)盐酸盐(58)的制备;
[0506]
向化合物57(1.80g,3.09mmol)的水(50ml)溶液中加入固体lioh
·
h2o(520mg,12.39mmol),并将反应混合物在室温下搅拌1小时,之后加入固体tcep
·
hcl(88mg,0.309mmol),将反应混合物在室温下搅拌2小时。反应完成后,其用4n hcl酸化,将残余物通过反相柱色谱纯化,得到为无色胶状固体的盐酸盐58(1.45g,91%):1h nmr(300mhz,cd3od)δ7.27(d,j=8.6hz,2h),6.98(d,j=8.6hz,2h),4.43-4.36(m,2h),4.21-4.07(m,2h),3.94-3.62(m,16h),3.57-3.42(m,2h);esi ms m/z 512[m+h]
+
。
[0507]
通用方法:所有试剂和溶剂购自aldrich chemical corp.chem-impex international inc.和tci chemical industry co.ltd。在bruker ac 400(400mhz处的1h nmr)或bruker ac 300(300mhz处的1h nmr)上获得nmr谱。除非另有说明,否则溶剂cdcl3、cd3od和dmso-d6购自aldrich或cambridge isotope laboratories。化学位移以相对于作为内标的四甲基硅烷(tms)的ppm报道。数据报告如下:化学位移、积分、多重性(s=单峰,d=双峰,t=三重峰,q=四重峰,br=宽,m=多重峰)和偶合常数(hz)。快速色谱法在装有硅胶柱(redi sep.rf,teledyne isco)或反相色谱柱(高性能c18 gold柱)的combiflash系统(combiflash rf,teledyne isco)上进行。在shimadzu lcms-2010ev质谱仪上获得esi质谱。在shimadzu prominence hplc系统上使用waters xterra ms c18 5μm 4.6x150mm分析柱在220nm检测(除非另有说明)获得hplc分析。通过tlc和lcms监测所有反应,通过hplc和lcms分析监测极性化合物反应。
[0508]
15.(s)-s-4-(2-氨基乙氧基)苄基2,6-二氨基硫代己酸酯的盐酸盐(64)的制备;
[0509]
方案15
[0510][0511]
(2-(4-(巯基甲基)苯氧基)乙基)氨基甲酸叔丁酯(61)的制备;
[0512]
向60(500mg,1.53mmol)在thf(50ml)、甲醇(10ml)和水(20ml)的混合物中的溶液中加入固体lioh
·
h2o(258mg,6.15mmol),然后加入tcep
·
hcl(880mg,3.06mmol),并将反应混合物在室温下搅拌2小时。反应完成后,其用4n hcl酸化,将残留物在饱和水(10ml)和
etoac(100ml)之间分配。分离有机层,水层用etoac(2
×
50ml)萃取。将合并的有机层经na2so4干燥,过滤并浓缩,得到为灰白色胶状固体的化合物61(380mg,88%):1h nmr(400mhz,cdcl3)δ7.23(d,j=8.4hz,2h),6.83(d,j=8.4hz,2h),4.97(br s,1h),4.00(t,j=5.2hz,2h),3.70(d,j=7.4hz,2h),3.54-3.50(m,2h),1.72(t,j=7.4hz,1h),1.45(s,9h);esi ms m/z 284[m+h]
+
。
[0513]
(s)-s-4-(2-((叔丁氧基羰基)氨基)乙氧基)苄基2,6-双((叔丁氧基羰基)氨基)硫代己酸酯(63)的制备;
[0514]
将化合物61(320mg,1.14mmol)和酸62(432mg,1.25mmol)溶于ch2cl2(100ml)中,在室温逐部分地加入edc
·
hcl(326mg,1.71mmol)和dmap(7.0mg,0.057mmol)。将反应混合物在室温下搅拌16小时,反应完成后,将溶液迅速用饱和nahco3水溶液(2
×
50ml)洗涤,随后用盐水(50ml)洗涤。将有机层经na2so4干燥并浓缩。将残余物通过柱色谱纯化,得到为灰白色固体的化合物63(620mg,89%):1h nmr(400mhz,cdcl3)δ7.18(d,j=8.8hz,2h),6.80(d,j=8.8hz,2h),5.11-4.98(m,2h),4.54(br s,1h),4.33(br s,1h),4.05(br s,2h),3.99(t,j=5.2hz,2h),3.53-3.45(m,2h),3.12-3.08(m,2h),1.88-1.78(m,1h),1.53-1.32(m,34h);esi ms m/z 612[m+h]
+
。
[0515]
(s)-s-4-(2-氨基乙氧基)苄基2,6-二氨基硫代己酸酯的盐酸盐(64)的制备;
[0516]
在室温下将化合物63(550mg,0.90mmol)溶于二噁烷(10ml)中的4n hcl,并将溶液在相同温度下搅拌2小时。除去溶剂后,将残余物通过反相柱色谱纯化,得到为吸湿性灰白色固体的盐酸盐564(190mg,50%):1h nmr(400mhz,cd3od)δ7.31(d,j=8.8hz,2h),6.96(d,j=8.8hz,2h),4.29-4.18(m,5h),3.35(t,j=4.8hz,2h),2.92(t,j=8.0hz,2h),2.06-1.90(m,2h),1.73-1.48(m,4h);1h nmr(400mhz,dmso-d6)δ8.70(br s,3h),8.28(br s,3h),8.06(br s,3h),7.29(d,j=8.8hz,2h),6.95(d,j=8.8hz,2h),4.22-4.15(m,5h),3.21-3.14(m,2h),2.74-2.66(m,2h),1.86-1.77(m,2h),1.61-1.49(m,2h),1.44-1.27(m,2h).esi ms m/z 312[m+h]
+
。
[0517]
16.(r)-s-4-(2-(2,6-二氨基己酰氨基)乙氧基)苄基硫代乙酸酯的盐酸盐(68)的制备;
[0518]
方案16
[0519][0520]
(r)-s-4-(2-(2,6-双((叔丁氧基羰基)氨基)己酰氨基)乙氧基)苄基硫代乙酸酯(67)的制备;
[0521]
将化合物65(1.00g,4.44mmol)和酸66(1.69mg,4.88mmol)溶于dmf(50ml)中,并用dipea(1.14g,8.88mmol)和hatu(2.53g,6.66mmol)处理。将反应混合物在室温下搅拌12小时。在减压下除去溶剂,将残余物溶于etoac(100ml)中,用饱和nahco3水溶液(2
×
100ml)快速洗涤溶液,然后用盐水(100ml)洗涤。将有机层经na2so4干燥并浓缩。将残余物通过柱色谱纯化,得到为灰白色固体的化合物67(1.60g,67%):1h nmr(300mhz,cdcl3)δ7.19(d,j=8.4hz,2h),6.81(d,j=8.4hz,2h),6.52(br s,1h),5.07(br s,2h),4.53(br s,1h),4.08-3.95(m,5h),3.68-3.60(m,2h),3.11-3.01(m,2h),2.33(s,3h),2.04(br s,1h),1.89-1.56(m,4h),1.53-1.19(m,24h);esi ms m/z 554[m+h]
+
。
[0522]
(r)-s-4-(2-(2,6-二氨基己酰氨基)乙氧基)苄基硫代乙酸酯的盐酸盐(68)的制备;
[0523]
在室温下将化合物67(300mg,0.542mmol)溶于二噁烷(10ml)中的4n hcl,并将该溶液在相同温度下搅拌2小时。除去溶剂后,将残余物通过反相柱色谱纯化,得到为吸湿性灰白色固体的盐酸盐68(150mg,60%):1h nmr(400mhz,cd3od)δ7.20(d,j=8.6hz,2h),6.85(d,j=8.6hz,2h),4.08-4.05(m,4h),3.85(t,j=6.6hz,1h),3.71-3.66(m,1h),3.59-3.54(m,1h),2.85-2.81(m,2h),2.33(s,3h),1.89-1.81(m,2h),1.68-1.62(m,2h),1.49-1.43(m,2h);1h nmr(400mhz,dmso-d6)δ8.75(t,j=5.4hz,1h),7.91(br s,5h),7.21(d,j=8.8hz,2h),6.87(d,j=8.8hz,2h),4.05-3.99(m,4h),3.69(t,j=6.8hz,1h),3.52-3.42(m,2h),2.69(t,j=7.4hz,2h),2.33(s,3h),1.72-1.66(m,2h),1.56-1.51(m,2h);esi ms m/z 354[m+h]
+
。
[0524]
17.(r)-2,6-二氨基-n-(2-(4-(巯基甲基)苯氧基)乙基)己酰胺(69)的盐酸盐制备;
[0525]
方案17
[0526][0527]
(r)-2,6-二氨基-n-(2-(4-(巯基甲基)苯氧基)乙基)己酰胺(69)的制备;
[0528]
将化合物67(350mg,0.632mmol)溶于甲醇(10ml)中,然后在室温下滴加于异丙醇(20ml)中的6n hcl,并将该溶液在相同温度下搅拌12小时。除去溶剂后,将残余物通过反相柱色谱纯化,得到为吸湿性灰白色固体的盐酸盐69(207mg,92%):1h nmr(300mhz,cd3od)δ7.24(d,j=8.6hz,2h),6.87(d,j=8.6hz,2h),4.09-4.03(m,2h),3.88(t,j=6.4hz,1h),3.71-3.55(m,4h),2.81(t,j=8.0hz,2h),1.92-1.82(m,2h),1.68-1.62(m,2h),1.50-1.39(m,2h);1h nmr(300mhz,dmso-d6)δ8.85(t,j=5.2hz,1h),8.30-8.04(m,6h),7.26(d,j=
8.6hz,2h),6.88(d,j=8.6hz,2h),4.02(br s,2h),3.77-3.67(m,3h),3.52-3.41(m,3h),2.80-2.68(m,3h),1.76-1.69(m,2h),1.60-1.51(m,2h),1.39-1.29(m,2h);esi ms m/z 312[m+h]
+
。
[0529]
18.(s)-s-4-(2-((r)-2-氨基-6-((叔丁氧基羰基)氨基)己酰氨基)乙氧基)苄基2,6-二氨基硫代己酸酯的盐酸盐(73)的制备;
[0530]
方案18
[0531][0532]
(r)-二-叔丁基(6-((2-(4-(巯基甲基)苯氧基)乙基)氨基)-6-氧代己烷-1,5-二基)二氨基甲酸酯(70)的制备;
[0533]
向67(700mg,1.26mmol)在thf(50ml)、甲醇(10ml)和水(20ml)的混合物中的溶液中加入固体lioh
·
h2o(212mg,5.06mmol),然后加入tcep
·
hcl(720mg,2.52mmol),将反应混合物在室温下搅拌2小时。反应完成后,其用1n hcl酸化,将残余物在饱和水(10ml)和etoac(100ml)之间分配。分离有机层,水层用etoac(2
×
50ml)萃取。将合并的有机层经na2so4干燥,过滤并浓缩,得到为灰白色固体的化合物70(500mg,78%):1h nmr(300mhz,cdcl3)δ7.23(d,j=8.6,2h),6.83(d,j=8.6,2h),6.58(br s,1h),5.10(br s,1h),4.55(br s,1h),4.08-3.97(m,3h),3.72-3.58(m,4h),3.11-2.98(m,2h),1.90-1.53(m,6h),1.51-1.17(m,25h);esi ms m/z 512[m+h]
+
。
[0534]
(s)-s-4-(2-((r)-2,6-双((叔丁氧基羰基)氨基)己酰氨基)乙氧基)苄基2,6-双((叔丁氧羰基)氨基)硫代己酸酯(72)的制备;
[0535]
将化合物70(500mg,0.97mmol)和酸71(372mg,1.07mmol)溶于ch2cl2(100ml)中,在室温下逐部分地加入edc.hcl(277mg,1.45mmol)和dmap(6.0mg,0.048mmol)。反应混合物在
d]咪唑-4-基)戊酰氨基)己酸(75)的制备;
[0543]
将化合物74(900mg,2.63mmol)和酸66(973mg,3.95mmol)溶于二噁烷(50ml)中,在室温下滴加6n naoh(10ml)。将反应混合物在室温下搅拌12小时,反应完成后,用etoac(100ml)稀释,将混合物用饱和nahco3水溶液(2
×
100ml)洗涤,随后用盐水(100ml)洗涤。将有机层经na2so4干燥并浓缩。将残余物通过柱色谱纯化,得到为灰白色固体的化合物75(700mg,56%):1h nmr(300mhz,cd3od)δ4.53-4.49(m,1h),4.34-4.30(m,1h),4.06(br s,1h),3.25-3.15(m,3h),2.97-2.91(m,1h),2.71-2.68(m,1h),2.23-2.17(m,2h),1.80-1.50(m,8h),1.45(s,13h);esi ms m/z 473[m+h].
+
[0544]
s-4-(2-((r)-2-((叔丁氧基羰基)氨基)-6-(5-((3as,4s,6ar)-2-氧代六氢-1h-噻吩并[3,4d]咪唑-4-基)戊酰氨基)己酰氨基)乙氧基)苄基硫代乙酸酯(77)的制备;
[0545]
将化合物75(700mg,1.48mmol)和胺76(367mg,1.63mmol)溶于dmf(50ml)中,并用dipea(381mg,2.96mmol)和hatu(843mg,2.22mmol)处理。将反应混合物在室温下搅拌12小时。在减压下除去溶剂,将残余物溶于etoac(100ml)中,并将溶液用饱和nahco3水溶液(2
×
100ml)快速洗涤,然后用盐水(100ml)洗涤。将有机层经na2so4干燥并浓缩。将残余物通过柱色谱纯化,得到为灰白色固体的化合物77(725mg,71%):1h nmr(300mhz,cd3od)δ7.18(d,j=8.4hz,2h),6.81(d,j=8.4hz,2h),7.11(br s,1h),6.09(br s,2h),5.36(br s,1h),5.24(br s,1h),4.46(t,j=4.4hz,1h),4.30(t,j=4.4hz,1h),4.06(br s,2h),4.01(t,j=5.2hz,2h),3.62(t,j=4.6hz,2h),3.23-3.11(m,3h),2.95-2.66(m,2h),2.32(s,3h),2.19(t,j=6.8hz,2h),1.82-1.57(m,8h),1.55-1.31(m,15h);esi ms m/z 540[m+h]
+
。
[0546]
s-4-(2-((r)-2-氨基-6-(5-((3as,4s,6ar)-2-氧代六氢-1h-噻吩并[3,4-d]咪唑-4-基)戊酰氨基)己酰氨基)乙氧基)苄基硫代乙酸酯(78)的制备;
[0547]
在室温下将化合物77(250mg,0.368mmol)溶于二噁烷(5ml)中的4n hcl,并将溶液在相同温度下搅拌2小时。除去溶剂后,将残余物通过反相柱色谱纯化,得到为吸湿性灰白色胶状物的盐酸盐78(150mg,70%):1h nmr(300mhz,cd3od)δ7.21(d,j=8.6hz,2h),6.87(d,j=8.6hz,2h),4.57-4.50(m,1h),4.38-4.31(m,1h),4.06(br s,4h),3.87-3.79(m,1h),3.77-3.50(m,4h),3.27-3.16(m,1h),3.14-3.05(m,2h),2.99-2.67(m,2h),2.31(s,3h),2.24-2.15(m,2h),1.90-1.77(m,2h),1.76-1.56(m,4h),1.55-1.31(m,6h);
[0548]1h nmr(300mhz,dmso-d6)δ8.68(br s,1h),8.11(br s,3h),7.72(br s,1h),7.21(d,j=8.6hz,2h),6.87(d,j=8.6hz,2h),6.38(br s,1h),4.32-4.26(m,1h),4.14-4.08(m,1h),4.07-3.96(m,4h),3.75-3.68(m,2h),3.56-3.46(m,3h),3.13-3.05(m,1h),2.99-2.90(m,2h),2.84-2.78(m,1h),2.59-2.54(m,1h),2.33(s,3h),2.03(d,j=7.2hz,2h),1.70-1.19(m,12h);esi ms m/z 616[m+h]
+
。
[0549]
20.(r)-2-氨基-n-(2-(4-(巯基甲基)苯氧基)乙基)-6-(5-((3as,4s,6ar)-2-氧代六氢-1h-噻吩并[3,4-d]咪唑-4-基)戊酰氨基)己酰胺盐酸盐(79)的制备;
[0550]
方案20
[0551][0552]
(r)-2-氨基-n-(2-(4-(巯基甲基)苯氧基)乙基)-6-(5-((3as,4s,6ar)-2-氧代六氢-1h-噻吩并[3,4-d]咪唑-4-基)戊酰氨基)己酰胺盐酸盐(79)的制备;
[0553]
将化合物77(475mg,0.699mmol)溶于甲醇(5ml)中,然后在室温下滴加异丙醇(15ml)中的6n hcl,并将该溶液在相同温度下搅拌12小时。除去溶剂后,将残余物通过反相柱色谱纯化,得到为吸湿性灰白色固体的盐酸盐79(260mg,69%):1h nmr(400mhz,cd3od)δ7.24(d,j=8.6hz,2h),6.87(d,j=8.6hz,2h),4.49-4.46(m,1h),4.30-4.27(m,1h),4.08-4.05(m,2h),3.80(t,j=6.4hz,1h),3.73-3.68(m,3h),3.59-3.53(m,1h),3.21-3.17(m,1h),3.09(t,j=7.2hz,2h),2.94-2.89(m,1h),2.69(d,j=12.4hz,1h),2.17(t,j=7.4hz,2h),1.84-1.35(m,12h);
[0554]1h nmr(300mhz,dmso-d6)δ8.68(t,j=5.4hz,1h),8.09(br s,3h),7.73(t,j=5.4hz,1h),7.25(d,j=8.4hz,2h),6.88(d,j=8.4hz,2h),6.36(d,j=11.2hz,2h),4.30(t,j=7.2hz,1h),4.13-3.97(m,3h),3.75-3.62(m,3h),3.57-3.42(m,2h),3.17-3.05(m,1h),2.97-2.91(m,2h),2.84-2.70(m,2h),2.57(d,j=12.4hz,1h),2.04(t,j=7.2hz,2h),1.69-1.42(m,6h),1.41-1.19(m,6h);esi ms m/z 574[m+h]
+
。
[0555]
21.(4-(2-氨基乙氧基)苯基)甲硫醇盐酸盐(84)的制备和s-(4-氨基乙氧基)苄基硫代乙酸酯盐酸盐(76)的制备;
[0556]
方案21
[0557][0558]
4-(2-((叔丁氧基羰基)氨基)乙氧基)苯甲酸乙酯(82)的制备;
[0559]
向于dmf(100ml)中的化合物80(8.00g,48.2mmol)的溶液中加入k2co3(26.6g,192.8mmol),并在室温下搅拌5分钟。向上述反应混合物中加入化合物21(21.5g,96.3mmol),将反应混合物在室温下搅拌20小时。将反应混合物用水(300ml)稀释,用etoac(3
×
300ml)萃取。将合并的有机层浓缩,将残余物通过柱色谱纯化,得到白色胶状的化合物82(10.2g,69%):1h nmr(400mhz,cdcl3)δ7.98(d,j=8.8hz,2h),6.90(d,j=8.8hz,2h),5.03(br s,1h),4.37-4.31(m,2h),4.07(t,j=5.2hz,2h),3.57-3.53(m,2h),1.45(s,9h),1.37(t,j=7.2hz,2h);esi ms m/z 310[m+h]
+
。
[0560]
(2-(4-(羟基甲基)苯氧基)乙基)氨基甲酸叔丁酯(83)的制备;
[0561]
在0℃向在thf(500ml)中的化合物82(10.0g,32.3mmol)的溶液中逐滴加入氢化铝锂(1m二乙醚溶液,66.4ml,32.3mmol)。将所得反应混合物在0℃下搅拌1小时,并在0℃用冰冷的水淬灭。将反应混合物用etoac(300ml)稀释并通过硅藻土垫过滤,并将硅藻土垫用etoac(2
×
300ml)洗涤。将滤液真空浓缩,通过柱色谱纯化,得到胶状固体的化合物83(6.10g,71%):1h nmr(400mhz,cd3cl)δ7.27(d,j=8.4hz,2h),6.86(d,j=8.4hz,2h),5.05(br s,1h),4.60(s,2h),3.99(t,j=5.2hz,2h),3.54-3.46(m,2h),2.03(br s,1h),1.44(s,9h);esi ms m/z 268[m+h]
+
。
[0562]
s-4-(2-((叔丁氧羰基)氨基)乙氧基)苄基硫代乙酸酯(60);sg-sur-g-48/51的制备
[0563]
在0℃向在ch2cl2(100ml)中的23(6.10g,22.8mmol)的溶液中加入et3n(2.76g,27.4mmol),然后加入甲磺酰氯(3.12g,27.4mmol),并在室温下搅拌2小时。将反应混合物用水(100ml)稀释,用ch2cl2(3
×
150ml)萃取。将合并的有机萃取液用盐水洗涤,经na2so4干燥,浓缩,得到黄色油状的甲磺酸化产物(8.20g,粗品),将其不经进一步纯化直接用于下一步骤:esi ms m/z 346[m+h]
+
。
[0564]
向上述在dmf(100ml)中的粗产物(8.20g,粗品)中加入ksac(6.70g,59.4mmol),并在室温下搅拌2小时。除去溶剂,将残余物在水(100.0ml)和etoac(100ml)之间分配。分离出
etoac层,水层用etoac(2
×
100ml)萃取。将合并的有机层浓缩,将残余物通过柱色谱纯化,得到为黄色固体的化合物1(4.80g,两步65%):1h nmr(400mhz,cd3od)δ6.91(s,2h),4.10(s,4h),3.99(t,j=5.7hz,4h),3.41(t,j=5.6hz,4h),2.31(s,6h),1.43(s,18);esi ms m/z 326[m+h]
+
。
[0565]
(4-(2-氨基乙氧基)苯基)甲硫醇盐酸盐(84)的制备;
[0566]
将化合物60(325mg,1.0mmol)溶于甲醇(5ml)中,然后在室温下逐滴加入异丙醇(1.0ml,6.0mmol)的6n hcl,并将溶液在相同温度下搅拌48小时。除去溶剂后,将残余物用etoac研磨,得到为吸湿性灰白色固体的盐酸盐84(190mg,87%):1h nmr(400mhz,cd3od)δ7.28(d,j=8.6,3.0hz,2h),6.93(d,j=8.6,3.0hz,2h),4.20(t,j=5.0hz,2h),3.69(s,2h),3.34(dd,j=5.0,4.0hz,2h)。
[0567]
400mhz(dmso-d6):δ7.90(brs,3h)7.28(d,j=8.8,3.0hz,2h),6.92(d,j=8.8,3.0hz,2h),4.13(t,j=5.2hz,2h),3.69(d,j=5.8hz,2h),3.19(t,j=5.2hz,2h),2.77(t,j=6.8hz,1h);esi ms m/z 184[m+h]
+
。
[0568]
s-4-(2-氨基乙氧基)苄基硫代乙酸酯盐酸盐(76)的制备;
[0569]
在室温下将化合物60(5.00g,15.4mmol)溶于二噁烷(20ml)中的4n hcl,并将溶液搅拌1小时。浓缩后,将残留物用mtbe研磨,得到为灰白色固体的盐酸盐76(3.6g,90%):1h nmr(400mhz,cd3od)δ7.23(d,j=8.5,3.0hz,2h),6.92(d j=8.5,3.0hz,2h),4.20(dd,j=5.2,4.8hz,2h),4.07(s,2h),3.34(dd,j=5.1,4.0hz,2h),2.31(s,3h);
[0570]
400mhz(dmso-d6):δ8.15(brs,3h)7.23(d,j=8.4,3.0hz,2h),6.91(d,j=8.4,3.0hz,2h),4.15(t,j=5.4hz,2h),4.06(s,2h),3.18(t,j=5.2hz,2h),2.33(s,3h);esi ms m/z 226[m+h]
+
。
[0571]
22.s-4-(2-(双((2s,3r,4r,5r)-2,3,4,5,6-五羟基己基)氨基)乙氧基)苄基硫代乙酸酯(25);sg-sur-g-05;alb187326的制备和(2r,2'r,3r,3'r,4r,4'r,5s,5's)-6,6'-((2-(4-(巯基甲基)苯氧基)乙基)氮烷二基)双(己烷-1,2,3,4,5-五醇)的盐酸盐(86)的制备;
[0572]
方案22
(3-氨基丙基)吡嗪-2-基)甲硫醇盐酸盐(94)的制备;
[0581]
方案23
[0582][0583]
5-(3-((叔丁氧基羰基)氨基)丙基)吡嗪-2-羧酸甲酯(89)的制备;
[0584]
在氩气下向化合物88(1.20g,7.64mmol)在无水thf(100ml)中的溶液中加入9-bbn(0.5m,在thf中,38ml,19.1mmol)。将反应混合物在室温下搅拌2小时后,在室温下加入化合物27(1.04g,6.11mmol)、pd(pph3)2cl2(311mg,0.38mmol)和2n na2co3水溶液(15ml)。将所得混合物再搅拌1小时。溶剂除去后,将残余物在etoac(200ml)和水(200ml)之间分配。分离水层,用etoac(2
×
200ml)萃取。将合并的有机萃取液用盐水洗涤,经na2so4干燥并真空浓缩。将粗产物通过柱色谱纯化,得到为棕色液体的化合物89(900mg,53%),其可直接用于下一步骤;esi ms m/z 296[m+h]
+
。
[0585]
(3-(5-(羟基甲基)吡嗪-2-基)丙基)氨基甲酸叔丁酯(90)的制备;
[0586]
在0℃向化合物89(900mg,3.05mmol)在etoh(100ml)中的溶液中加入硼氢化钠(400mg,15.25mmol)。将所得反应混合物在室温下搅拌3小时,并将残余物在etoac(200ml)和水(200ml)之间分配。分离水层,用etoac(2
×
200ml)萃取。将合并的有机萃取液用盐水洗涤,经na2so4干燥并真空浓缩。将粗产物通过柱色谱纯化,得到为棕色液体的化合物90(550mg,68%):1h nmr(400mhz,cd3cl)δ7.27(d,j=8.4hz,2h),6.86(d,j=8.4hz,2h),5.05(br s,1h),4.60(s,2h),3.99(t,j=5.2hz,2h),3.54-3.46(m,2h),2.03(br s,1h),1.44(s,9h);esi ms m/z 268[m+h]
+
。
[0587]
(5-(3-((叔丁氧基羰基)氨基)丙基)吡嗪-2-基)甲基甲磺酸盐(91)的制备;
[0588]
在0℃向90(260mg,0.97mmol)在ch2cl2(30ml)中的溶液中加入et3n(117mg,1.16mmol),然后加入甲磺酰氯(132mg,1.16mmol),并在室温下搅拌2小时。将反应混合物用水(100ml)稀释并用ch2cl2(2
×
50ml)萃取。将合并的有机萃取液用盐水洗涤,经na2so4干燥,浓缩,得到为黄色油状物的91(210mg,63%),其不经进一步纯化直接用于下一步骤:1h nmr(300mhz,cd3cl)δ8.62(s,1h),8.46(s,1h),5.33(s,2h),4.63(br s,1h),3.23-3.16(m,2h),3.11(s,3h),2.88(t,j=7.4,2h),2.00-1.91(m,2h),1.44(s,9h);esi ms m/z 346[m+h]
+
。
[0589]
(s)-((5-(3-((叔丁氧基羰基)氨基)丙基)吡嗪-2-基)甲基)硫代乙酸酯(92)的制备;
[0590]
向91(210mg,0.608mmol)在dmf(10ml)中的溶液中加入ksac(173mg,1.52mmol),并在室温下搅拌2小时。除去溶剂,将残余物在水(100.0ml)和etoac(100ml)之间分配。分离出etoac层,水层用etoac(2
×
100ml)萃取。将合并的有机层浓缩,残余物通过柱色谱纯化,得到为黄色固体的化合物92(140mg,71%):1h nmr(300mhz,cd3cl)δ8.52(s,1h),8.36(s,1h),4.65(br s,1h),4.22(s,2h),3.21-3.15(m,2h),2.82(t,j=7.4,2h),2.36(s,3h),1.97-1.87(m,2h),1.43(s,9h);esi ms m/z 326[m+h]
+
。
[0591]
s-((5-(3-氨基丙基)吡嗪-2-基)甲基)硫代乙酸酯盐酸盐(93)的制备;
[0592]
将化合物92(140mg,0.43mmol)溶于dcm(5ml)中,然后在室温下滴加于二噁烷(2ml)的4n hcl,并将溶液在相同温度下搅拌2小时。除去溶剂后,通过反相柱色谱纯化残余物,得到为吸湿性灰白色固体的盐酸盐93(80mg,71%):1h nmr(300mhz,cd3od)δ8.62(s,1h),8.51(s,1h),4.27(s,2h),3.04-2.92(m,4h),2.35(s,3h),2.12-2.07(m,2h);
[0593]1h nmr(300mhz,dmso-d6)δ8.56(s,1h),8.50(s,1h),8.10(br s,3h),4.24(s,2h),2.87-2.78(m,4h),2.36(s,3h),2.03-1.90(m,2h);esi ms m/z 226[m+h]
+
。
[0594]
(5-(3-氨基丙基)吡嗪-2-基)甲硫醇盐酸盐(34)的制备;
[0595]
将化合物92(600mg,1.84mmol)溶于甲醇(5ml)中,然后在室温下滴加于异丙醇(30ml)的6n hcl,并将该溶液在相同温度下搅拌12小时。除去溶剂后,将残余物通过反相柱色谱纯化,得到为吸湿性蓝绿色胶状物的盐酸盐94(290mg,85%):1h nmr(300mhz,cd3od)δ8.63(s,1h),8.48(s,1h),3.88(s,2h),3.05-2.92(m,4h),2.15-2.05(m,2h);
[0596]1h nmr(300mhz,dmso-d6)δ8.61(s,1h),8.48(s,1h),7.93(br s,3h),3.85(d,j=8.0hz,2h),3.03(t,j=8.0hz,1h),3.05-2.92(m,4h),2.15-2.05(m,2h);esi ms m/z 184[m+h]
+
。
[0597]
dtnb测定:含硫醇化合物,诸如本专利所述的单硫醇粘液溶解剂可与dtnb(5,5-二硫代双(2-硝基苯甲酸))反应,导致其s-s单键断裂。dtnb测定在我们对单硫醇化合物的分析中具有双重目的,因为其可(1)用于确定粘液溶解化合物的“活性浓度”(反应中化合物能够裂解s-s键的量)和(2)评价化合物能够裂解s-s键的速率。当裂解时,所得的化合物ntb(2-硝基-5-硫代苯甲酸酯)是在412nm的吸光度(abs
412
)下用分光光度法测量的有色产物。使用ntb的摩尔消光系数和测量的最大abs
412
,可计算与我们的粘液溶解剂反应的dtnb的摩尔量。单硫醇粘液溶解化合物以1:1的化学计量比与dtnb反应(即当1摩尔dtnb与1摩尔单硫醇还原剂反应时,产生1摩尔ntb)。为了确定化合物的活性浓度,将终浓度为22.5μm的粘液溶解还原剂与过量的dtnb(100μm)在1ml 50mm tris-hcl缓冲液(ph 7.5)中合并。测量最大abs
412
,然后用于计算活性浓度。如果观察到的活性浓度与预期的活性浓度的差异超过5%,则相应地调整添加至每个反应中的化合物的量以动力学测试下一步骤中的还原速率。为了测定粘液溶解还原活性的速率,将22.5μm活性还原剂与45μm dtnb在ph范围(6.0-7.5)内的1ml 50mm tris-hcl缓冲液中合并。在添加化合物后实时记录abs
412
,并使用2阶动力学参数计算速率。结果列于表1。
[0598]
表1.本发明化合物相较于nac的还原dtnb的动力学反应速率
[0599][0600]
前药dtnb测定:该测定被设计用于确定添加至我们的单硫醇粘液溶解化合物上的前药封端(prodrug caps)是否可以从分子中裂解出来。如果是这样,结果将是能够还原dtnb中s-s键的“活性”化合物。类似于上述dtnb测定的活性浓度版本,将单硫醇前药以22.5μm的终浓度与过量dtnb(最终100μm)在1ml 50mm tris-hcl缓冲液(ph 7.5)中混合,并测量最大abs
412
。在没有纯化的水解酶(例如猪肝酯酶;sigma)的情况下,观察到的dtnb裂解的量是前药化合物上的封端的稳定性的指标(即如果前药在溶液中保持完整,则abs
412
=0)。然后,将5μl酯酶(购买作为随后离心分离酯酶然后在分析前溶于反应缓冲液中的硫酸铵悬浮液)与过量的dtnb(最终100μm)和22.5μm前药在1ml 50mm tris-hcl缓冲液(ph7.5)中合并。可通过经酯化酶释放的单硫醇分子在abs
412
下通过dtnb裂解,实时可视化前药封端的裂解。结果列在表2中。
[0601]
表2.添加和未添加酶的前药的还原
[0602][0603]
平行人造膜通透性(pampa)测定:该测定法测量小分子穿过人造磷脂膜的通透性,以帮助预测体内药物通透性。首先,在pbs中制备500μm原液,并通过dtnb测定(如上所述)测试其以测定化合物活性浓度。然后,分别向供体板和受体板(bd gentest
tm
预涂pampa板系统)(bd gentest
tm pre-coated pampa plate system)加入300μl的500μm化合物原液和200μl pbs。然后将受体板置于供体板的顶部,使其在室温下孵育5小时。孵育后,分离供体和受体平板。对于每个反应,从受体和供体平板上取样品,将其与dtnb合并,然后在分光光度计(最大abs
412
)上读数。对于前药样品,每孔加入1μl纯化的酯酶(猪肝酯酶;sigma),并将样品在室温下孵育5分钟以使裂解前药封端,然后加入dtnb。对于每个样品使用在供体和受体板中鉴定的化合物的浓度,按照bdgentest
tm
预涂pampa板系统手册中概述的方程式计算化合物渗透率参数。
[0604]
粘蛋白琼脂糖凝胶免疫印迹:在完成处理期后,用10倍过量的n-乙基马来酰亚胺淬灭用我们的单硫醇粘液溶解化合物处理的各种含粘蛋白的底物(例如,唾液、收获的hbe粘液和疾病痰),以烷基化任何剩余的活性化合物,并防止进一步的粘蛋白还原。将10倍浓缩的样品加样缓冲液稀释到每个样品(1x tae/5%甘油/0.1%sds/溴酚蓝)中。使用由1x tae/0.1%sds组成的缓冲系统通过在0.9%琼脂糖凝胶上电泳来分析样品(50μg)。将琼脂糖凝胶在含有10mm dtt的4xssc(0.6m nacl,60mm柠檬酸三钠脱水剂)中浸泡15分钟,然后
通过真空印迹装置将样品从凝胶转移至硝酸纤维素膜上。使用针对muc5b的多克隆抗体,在licor odyssey成像检测系统上可视化未还原和还原的muc5b。
[0605]
bip诱导:在缓冲至ph 7.5的50mm tris-hcl中制备还原剂。将每种化合物溶液(15μl的10mm化合物)加入到原代hbe的顶端表面,持续24小时。然后在补充有蛋白酶抑制剂混合物(roche)和1mm苯基甲基磺酰基氟化物的ripa缓冲液(50mm tris-hcl(ph8.0)/150mm nacl/1.0%np-40/0.5%脱氧胆酸钠/0.1%sds)中裂解细胞。将样品归一化以含有相同的蛋白质总量,然后加入2x sds样品缓冲液(100mm tris-hcl(ph6.8)/4%sds/0.05%溴酚蓝/20%甘油)。通过在10%sds-page凝胶上电泳来分析样品(20μg),并将其转移至硝酸纤维素膜上。使用针对bip的多克隆抗体和licor odyssey成像检测系统来可视化bip水平。将thapsigargin(tg,2.5μm)和dtt作为bip诱导的阳性对照。
[0606]
il-8分泌:在缓冲至ph7.5的50mmtris-hcl中制备还原剂。将每种化合物溶液(15μl的10m m化合物)加入至原代hbe的顶端表面。
[0607]
使用thermoscientific人il-8elisa试剂盒,在剂量后24小时从底外侧hbe培养基测量il-8分泌。将样品以1:50稀释,然后将50μl的每种稀释物加入至elisa板中。按照试剂盒手册中所述的标准反应条件以一式两份完成样品。在450nm处测量吸光度。使用从具有已知量的人il-8的标准产生的标准曲线来计算未知样品中的il-8量。使用从囊性纤维化痰纯化的粘脓性物质(smm)的上清液作为il-8分泌的阳性对照。
[0608]
使用这些测定的数据汇总于在图2-9中并如下详细讨论如下:
[0609]
如图2所示,提供了化合物76相对于nac的增强的粘蛋白还原能力。新型粘液溶解剂包含若干特征以产生相对于目前可用的药剂具有有利性能的治疗剂。唯一被批准用于吸入的粘液溶解的nac的一个限制是其非常缓慢的还原动力学,需要非常高的药物浓度和长孵育时间。化合物76,一种新型粘液溶解的战斗部(warhead),显示相对于nac的显著提高的动力学。如通过琼脂糖凝胶免疫印迹所示,相对于nac(50mm),等摩尔浓度的76相比nac产生显著更快和更完全的唾液muc5b的还原。此外,76在低至1/5的浓度(10mm)下相对于nac产生更大的muc5b还原。因此,与nac相比,76表现出更有活性的还原剂的性质。
[0610]
如图3所示,可以实现化合物76的修饰而不损失还原活性。如通过衍生物86、69和47所举例说明的,76的功能性取代保留了76的还原能力。如图所示,所有化合物在还原muc5b方面显著高于nac的活性(如通过免疫印迹上的高分子量muc5b条带的损失指示的)。
[0611]
在图4中,化合物86相对于本领域已知的化合物的还原活性。通过琼脂糖凝胶免疫印迹,86证明了相对于nac和本领域已知的另一种药剂(wo2014153009中描述的化合物95)的更优的muc5b还原活性。只有86在30mm下产生完全muc5b还原,在3mm下产生部分还原;而其他化合物仅在测试的最高浓度(30mm)下产生部分muc5b还原。
[0612][0613]
图5显示了化合物76取代物的优点。nac作为肺粘液溶解剂的主要限制是有限的气道表面药物保留(mucomyst产品说明书描述了20分钟的肺表面半衰期)。如图所示,如使用pampa测定法所测量的,76战斗部是相对亲脂的和膜可渗透的。然而,取代的76分子,诸如86
和85(其相对于nac保持76的增强的活性)是更加极性和不太亲脂的,这导致差的膜通透性。因此,数据预测86和85将有利地显示在肺表面上的提高的化合物保留。
[0614]
图6显示用于评估细胞渗透和促炎症作用的综合测定。将化合物在极化hbe细胞的顶端表面上以10mm孵育24小时,并测量(a)bip蛋白或(b)il-8分泌的诱导。bip蛋白的诱导是内质网(er)应激的标志物,其可通过蛋白质合成中二硫键形成的抑制而引起。由于预测86难以渗透细胞,所以预计该化合物不会到达er,因此不诱导bip表达。图a:86和69相对于载体对照(虚线)不诱导bip表达,而可渗透还原剂(dtt)和thapasgargin(tg)则诱导bip表达。因此,数据表明当加入至顶端区室时,86和69未被显著内化在细胞中。图b:为了评估化合物的潜在促炎特性,在24小时化合物孵育后测量il-8分泌。显著地,86没有产生任何促炎症作用的证据,如通过il-8无变化所证明的。然而,69以及阳性对照(smm)确实增加了il-8分泌。由于86和69共有共同的战斗部,76(其没有增加il-8分泌),我们得出结论,69的取代物,而不是86,负责该作用。
[0615]
图7显示了86的前药。85是86的硫代乙酸酯衍生物。重要的是,前药部分使85无臭和无活性(直至化合物被代谢活化)。图显示了dtnb测定中的86,其快速裂解dtnb(左图)。相反,85没有裂解dtnb(中图),表明该化合物是无活性的。然而,酯酶的添加将85代谢转化成86,允许其水解dtnb(右图)。
[0616]
图8显示copd痰中85的代谢活化。粘液溶解剂前药的一个重要方面是它们必须在肺表面上的粘液中活化。为了评估代谢活化和动力学,将86(活性代谢物)和85(前药)从copd患者的自发诱导的痰中孵育。如图所示,86迅速还原痰中的muc5b,如通过琼脂糖凝胶免疫印迹法评估的。85同样还原muc5b,但相对于86具有延迟的动力学(这反映了对85至86的酶促代谢的需求)。重要的是,这些数据表明人的痰含有活化含硫酯化合物诸如85所需的酶。
[0617]
图9显示了原代人支气管上皮(hbe)培养物上的粘液还原。在将粘液溶解剂加入至hbe培养物的顶端表面后指定的时间上通过蛋白质印迹评估粘蛋白还原动力学。86产生了在给药后24小时基本上恢复的粘液的快速还原。85表现出较慢的还原动力学(例如1-2小时),但潜在地较长的作用持续时间(例如24小时)。重要的是,数据表明,具有粘液的人气道上皮可将85前药代谢为活性86粘液溶解剂。
[0618]
如上在整个本申请中引用的每个参考文献通过参考并入本文。在前述描述与参考文献之间发生冲突的情况下,以本文提供的描述为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1