一种近红外氮杂氟硼二吡咯染料及其微波法合成方法与流程
本发明涉及有机合成技术领域,具体涉及一种近红外氮杂氟硼二吡咯荧光染料及其微波合成方法。
背景技术:
近红外氮杂氟硼二吡咯染料,作为一种新型的荧光染料,因其具备优异的光学性质和能作为光敏剂的特性,近年来已经成为光功能材料研究的热点,在生物医学领域具有非常广阔的应用前景(J.Am.Chem.Soc.2004,126,10619-10631)。四芳基氮杂二吡咯甲川类化合物与BF2络合,阻止了与吡咯环相连的桥氮键发生异构,得到刚性平面更好的近红外氮杂氟硼二吡咯染料,显现出优异的光学性质:荧光团发射和吸收波长都在近红外区域;更高的摩尔吸光系数(70000-80000M-1cm-1)(Org.Lett.,2006,8,3493-3496)和荧光量子产率(0.3-0.6);优异的光稳定性,受环境、溶液pH影响较小(Org.Lett.,2013,15,3392-3395)等。荧光波长在近红外区域,具备更高的生物组织穿透性,能够有效地消除细胞自体荧光干扰(Org.Lett.,2008,10,4771-4774),降低在观察过程中对细胞或组织的损伤(Org.Biomol.Chem.,2010,8,522-525)。
除了具备优异的光学性质,这类染料还具备光敏剂的特性,用于癌症的治疗和早期诊断(RSC Adv.,2012,2,11169-11183)。光敏剂在生物体内,对肿瘤组织和正常组织细胞亲和力不同,导致滞留时间不同,使其被动分散聚集在肿瘤中, 利用特定波长光照射,激发光敏剂从基态S0跃迁到激发态S1,由于能量的系间跨越(ISC),导致激发态S1到激发态三重态T1,能量由T1转移到细胞中的基态氧,会产生具有细胞毒性的单线态氧,导致细胞凋亡(Chem.Commun.,2002,1862-1863;J.Am.Chem.Soc.2005,127,16360-16361)。因为光照只对受照射区域产生影响,使得PDT(Photodynamic therapy光动力学疗法)对于癌症的治疗具有极高的选择性(J.Med.Chem.2010,53,7337-7343)。优异的光学性质和作为光敏剂的特性使得这类染料在生物检测领域具有极大的潜在应用价值。
从这类化合物第一次被报道(J.Chem.Soc.1943,590-596)以来,Donal F.O’Shea课题组不断优化合成条件,相继制得不同功能的氮杂氟硼二吡咯染料,推动了这类荧光染料的研究进展(J.Am.Chem.Soc.2011,133,19618-19621;Int.J.Cancer 2011,130,705-715;J.Org.Chem.2012,77,9304-9312;Org.Lett.,2013,15,3392-3395)。目前常用合成氮杂氟硼二吡咯化合物的路线如下:
以α,β-不饱和酮为原料,经硝基甲烷亚硝化,采用常规加热法以无水乙醇为溶剂与醋酸铵反应,合成四芳基氮杂二吡咯甲川类化合物,与三氟化硼络合得到氮杂氟硼二吡咯分子。在合成四芳基氮杂二吡咯甲川类化合物过程中,利用常规加热法存在反应时间长(24h–72h),产率较低(12%-47%)的缺陷,使得整个合成路线完成时间长,产率低。
技术实现要素:
针对现有技术中存在的不足,本发明提供了一种新的近红外氮杂氟硼二吡咯染料化合物及其微波法合成方法,其目的在于弥补现有合成该类化合物的方法中 存在的一些不足,相比于传统的常规加热回流法,能够缩短反应时间、提高反应产率、减少副产物。
技术解决方案
一种新的近红外氮杂氟硼二吡咯染料,结构式如下:
本发明还进一步提供了一种所述近红外氮杂氟硼二吡咯染料的微波合成方法,合成路线如下:
上述合成方法包含以下步骤:
步骤一:对羟基苯乙酮和对氰基苯甲醛在碱性条件下反应,得到α,β-不饱和酮1。将1:1摩尔比的对羟基苯乙酮和对氰基苯甲醛溶于无水乙醇,加入3倍对羟基苯乙酮摩尔量的氢氧化钾,室温搅拌过夜,再加3倍对羟基苯乙酮摩尔量的氢氧化钾,继续室温搅拌5h,反应结束。抽滤,滤液用3mol/L盐酸调节溶液pH=4.0,有沉淀析出,抽滤得到化合物1为淡黄色固体。
氢氧化钾加入量和pH调节至关重要,如若条件不合适,析出的固体可能不是所要产物,或者很大程度上降低产率。
步骤二:α,β-不饱和酮与硝基甲烷、二乙胺(三者摩尔量比为1:10:5), 发生Michael加成反应进行亚硝化,得到化合物2。将化合物1加热溶于无水乙醇,加入二乙胺和硝基甲烷,加热回流12h。反应结束,用3mol/L盐酸调节溶液pH=3.0,用乙酸乙酯萃取,有机层干燥,真空去除溶剂,柱层析分离提纯,得到化合物2为白色固体。
步骤三:利用微波法,化合物2与醋酸铵反应生成化合物3。化合物2溶于无水乙醇,加入35倍化合物2摩尔量的醋酸铵,将混合液加入微波反应容器中,在95℃下微波反应6h。有固体析出,抽滤,固体用0℃的无水乙醇洗涤,得到化合物3为具有金属光泽蓝紫色固体。
步骤四:化合物3与三氟化硼(摩尔比为1:17)络合得到化合物4。化合物3溶于重蒸的甲苯,加入三氟化硼乙醚溶液(48%BF3)和三乙胺,80℃下搅拌过夜,络合得到所述的近红外氮杂氟硼二吡咯染料4,为红色具有金属光泽固体。
后处理如下:将搅拌过夜后的混合物冷却至室温,真空去除溶剂,用乙酸乙酯溶解残留物,水洗3次,有机相用无水硫酸钠干燥,去除溶剂得到深灰色固体,用二氯甲烷重结晶。
该近红外氮杂氟硼二吡咯化合物上的氰基可以使该类染料波长在很大程度上发生红移,使吸收和发射波长位于近红外区域,具备更高的生物组织穿透性,有效地消除细胞自体荧光干扰,此外降低在观察过程中对细胞或组织的损伤。羟基的引入,针对肿瘤细胞的pH在酸性范围,可以使该化合物成为一种pH敏感的分子探针,对肿瘤细胞进行荧光成像。
本发明微波法合成一种近红外氮杂氟硼二吡咯染料,也适用于其他近红外氮杂氟硼二吡咯染料的合成,优点在于:微波法相比于常用的加热回流合成法,对于合成该类荧光染料,可以大大缩短反应时间、减少副产物、提高反应产率。
附图说明
图1为实施例1所合成化合物1的1H NMR。
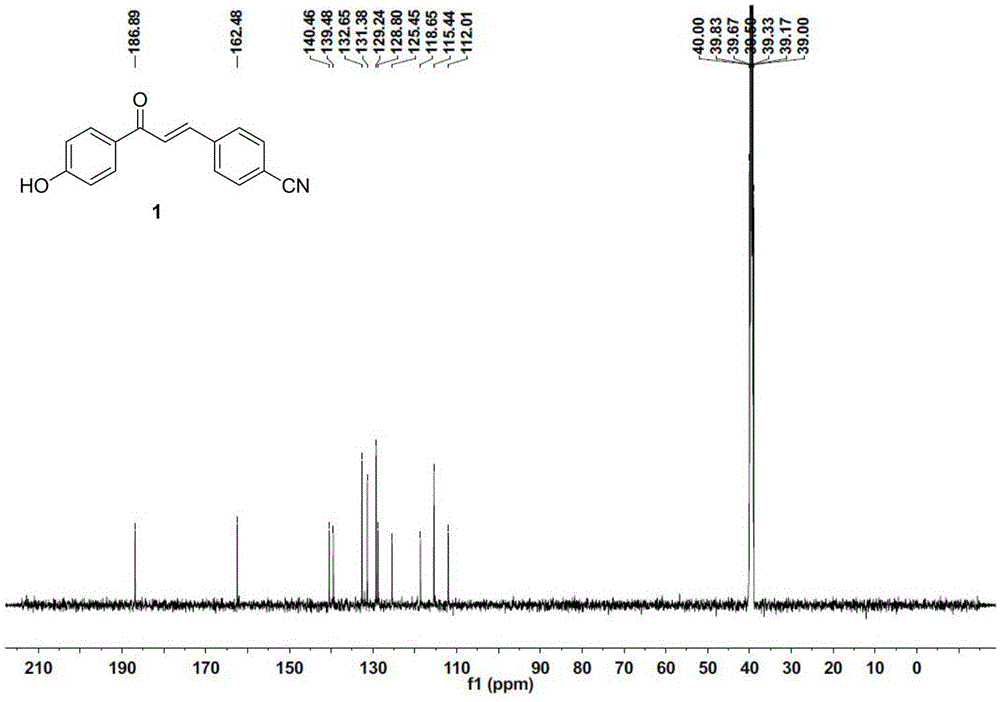
图2为实施例1所合成化合物1的13C NMR。
图3为实施例1所合成化合物2的1H NMR。
图4为实施例1所合成化合物2的13C NMR。
图5为实施例1所合成化合物3的1H NMR。
图6为实施例1所合成化合物3的13C NMR。
图7为实施例1所合成化合物4的1H NMR。
图8为实施例1所合成化合物4的13C NMR。
图9为实施例1所合成化合物4的19F NMR。
图10为实施例1所合成化合物3的TIC图和高分辨质谱图。从图中可以看出,得到的化合物3纯度高,杂质少。
图11为实施例1所合成化合物4的紫外可见吸收光谱(10μM)。最大吸收波长为745nm。
图12为实施例1所合成化合物4在最佳激发波长745nm(10Μm)下的荧光发射光谱,最大发射波长在779nm。说明该染料的吸收和发射波长都在近红外区域。
图13为本发明提供的一种新的近红外氮杂氟硼二吡咯染料的化学结构式。
具体实施方式
以下以具体实施例来说明本发明的技术方案,但本发明的保护范围不限于此:
以下实施例中微波反应所用的微波反应器为Microwave Synthesis Reactor:Monowave 300(Anton Paar)。
实施例1
近红外氮杂氟硼二吡咯染料化合物4的合成:
1.合成化合物1:
对羟基苯乙酮(1.36g,10mmol)和对氰基苯甲醛(1.31g,10mmol)溶于50mL无水乙醇,加入KOH(560mg,30mmol),室温搅拌过夜,然后加入KOH(560mg,30mmol),室温搅拌5h,抽滤,滤液用3mol/L盐酸调节溶液pH=4.0,有沉淀析出,抽滤,滤饼用3mol/L盐酸洗涤,得到化合物1为淡黄色固体粉末(1.80g,产率72.3%)。1H NMR(500MHz,DMSO-d6)δ10.48(s,1H),8.10-8.05(m,5H),7.92(d,J=5.0Hz,2H),7.72-7.69(d,J=15.0Hz,1H),6.91-6.90(d,J=5.0Hz,2H).13C NMR(126MHz,DMSO-d6)δ186.89,162.48,140.46,139.48,132.65,131.38,129.24,128.80,118.65,115.44,112.01.HRMS(ESI+)calcd for C16H11NO2[M+H]+250.0863,found 250.0848.
2.合成化合物2:
化合物1(120mg,0.48mmol)溶于25mL无水乙醇,加热回流至固体溶解。加入硝基甲烷(0.25mL,4.8mmol)和二乙胺(0.24mL,2.4mmol),回流12h,冷却至室温,用3mol/L盐酸调节溶液pH=3.0。乙酸乙酯萃取,有机层用无水硫酸钠干燥,真空去除溶剂,柱层析分离提纯(正己烷/乙酸乙酯=1:1),得化合物2为白色固体(90mg,产率60.5%)。1H NMR(500MHz,DMSO-d6)δ10.40(s,1H),7.82-7.77(m,4H),7.61(d,J=5.0Hz,2H),6.84-6.82(m,2H),5.04-4.90(m,2H),4.13-4.07(m,1H),3.54-3.41(m,2H).13C NMR(126MHz,DMSO-d6)δ194.98,162.28,146.15,132.31,130.50,129.05,127.84,118.64,115.22,110.01,79.02.HRMS(ESI+)calcd for C17H14N2O4[M+H]+311.1026,found 311.1051.
3.合成化合物3:
化合物2(90mg,0.29mmol)溶于5mL无水乙醇,加入醋酸铵(777m g,10.1mmol),将混合液加入微波反应专用玻璃管中,在95℃下微波反应6h,TLC监测原料完全反应。有固体析出,抽滤,固体用冷的乙醇洗涤,得到具有金属光泽蓝紫色固体3(40mg,产率52.6%)。1H NMR(500MHz,DMSO-d6)δ10.32(s,2H), 8.23(d,J=10.0Hz,4H),7.99(d,J=10.0Hz,4H),7.93(d,J=10.0Hz,4H),7.72(s,2H),7.01(d,J=10.0Hz,4H).13C NMR(126MHz,DMSO-d6)δ160.46,154.82,148.87,138.54,137.77,132.41,128.85,128.82,122.06,119.11,116.95,116.56,110.06.HRMS(ESI+)calcd for C34H21N5O2[M+H]+532.1768,found 532.1772。
4.合成氮杂氟硼二吡咯染料化合物4:
化合物3(15mg,0.028mmol)溶于10mL重蒸甲苯,搅拌10min,加入三乙胺(0.1mL,0.28mmol),混合液加热至80℃,加入三氟化硼乙醚溶液(0.2mL,0.48mmol),立刻有大量深蓝色固体析出,保持80℃反应过夜,冷却至室温,真空去除溶剂,用乙酸乙酯溶解残留物,水洗3次,有机相用无水硫酸钠干燥,去除溶剂得到深灰色固体,用二氯甲烷重结晶得到红色金属光泽固体粉末4(15mg,产率92.6%)。1H NMR(500MHz,DMSO-d6)δ10.60(s,2H),8.32-8.31(d,J=5.0Hz,4H),8.13-8.07(dd,J=10.0,5.0Hz,8H),7.78(s,2H),6.97-6.96(d,J=5.0Hz,4H).13C NMR(126MHz,DMSO-d6)δ161.34,157.60,144.46,138.94,136.03,132.52,132.33,129.23,121.32,121.13,118.77,115.99,111.27.19F NMR(470MHz,CDCl3)δ-131.05(q,J=28.2Hz,BF2).HRMS(ESI+)calcd for C34H20BF2N5O2[M–CN+H]+554.1726,found 554.1733.
- 还没有人留言评论。精彩留言会获得点赞!