一种与Aβ斑块具有亲和力的荧光化合物及制备与应用的制作方法
本发明属于特异性分子识别诊断试剂领域,具体涉及一种与aβ斑块具有亲和力的荧光化合物及制备与应用。
背景技术:
阿尔茨海默症(alzheimer’sdisease,ad)是一种神经退行性疾病。根据2016年一项最新调查数据显示,目前全球范围内有超过4700万ad病人。而中国已提前进入老龄化社会,调查显示我国已有超过800万ad患者,数量居世界之首,且患病人数以每年30万以上的速度递增。ad不但严重影响老年人的身体健康,而且给社会和家庭带来沉重的负担。目前,临床上可用于治疗ad的药物不能延缓、终止或者逆转ad病情的发展,只能部分改善临床症状。而且,由于ad病因复杂,确切的发病机理目前尚不清楚,临床上主要通过评价患者的认知能力损伤来诊断,确诊患者多已进入病程的中晚期而延误了治疗。缺乏有效的检测手段已成为ad早起诊断和治疗的重大障碍。
在ad患者脑中,β-淀粉样蛋白(amyloid,aβ)在脑中沉积是ad最为显著的病理性标志之一。aβ斑块在ad发病前的数十年已开始出现,是ad最早的神经组织退化标志和重要病理学特征。近年来,aβ斑块形成的预防和逆转成为治疗ad的目标之一,抑制aβ斑块在脑内产生和蓄积的药物和疗法得到了广泛研究。
除了ad外,aβ斑块也存在于其他疾病中,例如脑淀粉样血管病、淀粉样心肌病、淀粉样多发性神经病、系统性老年淀粉样变和伴有淀粉样变的遗传性脑出血等。因此,研发具有特异性结合aβ斑块的aβ分子探针引人关注。利用aβ分子探针和分子成像技术进行aβ斑块的显像,可实现无创的、实时的aβ斑块的体内示踪和检测,进而为ad等疾病患者进行早期诊断、疗效检测以及治疗药物的研究等提供极大便利。
过去的数年间,已有较多放射性aβ分子探针进入临床试验阶段,并有相关pet成像试剂上市,但是放射性显像方法的应用也受一些因素限制,比如放射性显像剂所发出的射线对人体具有一定的辐射损伤、需要医疗机构配套有生产放射性核素的回旋加速器、放射性显像剂需专业技术人员标记配制等。相比较之下,光学成像具有安全无放射性、数据采集时间短以及成本低廉等诸多优势,近年在医学诊断等中的应用受到广泛重视。尤其是近红外荧光成像技术,因其背景荧光干扰较低,在生物组织中穿透力强,因此,研发对aβ斑块具有亲和力的近红外荧光显像剂(分子探针),将具有重要的科学意义和实际价值。
技术实现要素:
针对以上现有技术存在的缺点和不足之处,本发明的首要目的在于提供一种与aβ斑块具有亲和力的荧光化合物。
本发明的另一目的在于提供上述与aβ斑块具有亲和力的荧光化合物的制备方法。
本发明的再一目的在于提供上述与aβ斑块具有亲和力的荧光化合物的应用。
本发明目的通过以下技术方案实现:
一种与aβ斑块具有亲和力的荧光化合物,所述荧光化合物具有式(i)所示的结构通式:
其中,r为
上述荧光化合物的制备方法,包括以下制备步骤:
(1)将2,3,3-三甲基吲哚和碘甲烷加入到乙醇混合搅拌均匀,在耐压容器中加热到60~100℃反应,反应完成后冷却至室温析出固体,抽滤,得到中间体1:
(2)将dmf和二氯甲烷混合均匀,冰浴并氮气保护,然后滴加三氯氧磷,搅拌20~40min后加入环己酮,升温至50~100℃回流反应,反应完成后冷却至室温,将反应液加入冰水中,析出固体,抽滤,得到中间体2:
(3)将中间体1和中间体2溶于有机溶剂中,升温至80~130℃回流反应,反应完成后旋蒸除去溶剂,柱层析分离得到中间体3:
(4)将中间体3和丙二腈溶于有机溶剂中,加入哌啶或碳酸钾甲醇饱和溶液,室温搅拌析出固体,抽滤,得到与aβ斑块具有亲和力的荧光化合物dcsc-1;或将中间体3和氰基乙酸乙酯溶于有机溶剂中,加入哌啶或碳酸钾饱和溶液,室温搅拌反应后,柱层析分离,得到与aβ斑块具有亲和力的荧光化合物dcsc-7。
优选地,步骤(3)中所述的有机溶剂为正丁醇与甲苯的混合溶剂。
优选地,步骤(4)中所述的有机溶剂为甲醇。
优选地,步骤(4)中所述中间体3与丙二腈或氰基乙酸乙酯的摩尔比为1:(5~15)。
上述制备方法的合成路线图如图1所示。
上述与aβ斑块具有亲和力的荧光化合物在制备aβ斑块显像诊断组合物中的应用,所述组合物包括式(i)所示的荧光化合物与药学上可接受的载体,所述“药学上可接受的载体”包括各种赋形剂和稀释剂。所述“药学上可接受的载体”可包括液体,如水、生理盐水、甘油或乙醇。
上述与aβ斑块具有亲和力的荧光化合物在制备aβ斑块诊断试剂或与aβ沉积相关疾病的治疗药物中的应用。
进一步地,所述与aβ沉积相关疾病是指阿尔茨海默症或脑淀粉样血管病。
本发明的荧光化合物具有如下优点及有益效果:
本发明的荧光化合物,其结构中的给电子基团和吸电子基团形成了推-拉电子作用的共轭结构,并通过碳碳双键增加跃迁的共轭体系,使化合物分子产生的荧光向近红外区移动,同时本类化合物均具有良好的荧光性质,发射波长达到了近红外区。所得荧光化合物在体外实验表现出对aβ斑块具有亲和力,能够成功用于aβ斑块的近红外荧光显像,具有安全无放射性、成本低廉、背景荧光干扰较低、在生物组织中穿透力强等优点。
附图说明
图1是本发明荧光化合物(i)的合成路线图;
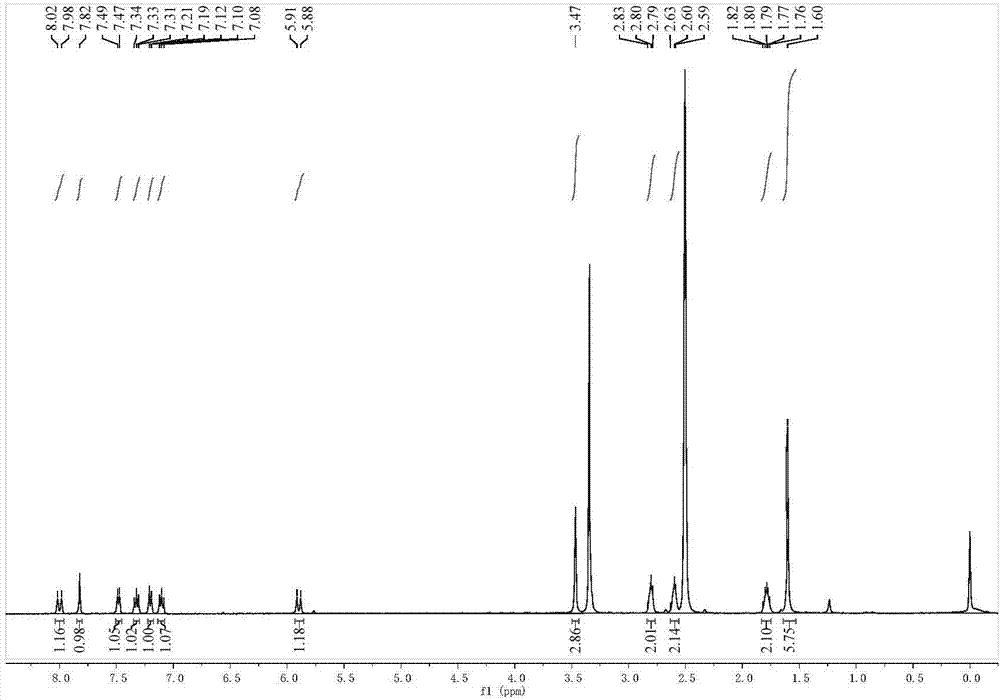
图2是本发明实施例所得荧光化合物dcsc-1的1h-nmr谱图;
图3是本发明实施例所得荧光化合物dcsc-1的13c-nmr谱图;
图4是本发明实施例所得荧光化合物dcsc-7的1h-nmr谱图;
图5是本发明实施例所得荧光化合物dcsc-7的13c-nmr谱图;
图6是本发明所得荧光化合物dcsc-1和dcsc-7制备的探针(i)与aβ1-42聚集体混合前后的荧光发射光谱图;
图7是本发明所得荧光化合物dcsc-1溶液探针由尾静脉注入ad转基因小鼠和正常小鼠后60min内不同时间点脑部活体成像图。
具体实施方式
下面结合实施例及附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。
以下实施例对于未特别注明的参数,可参照常规技术进行。核磁谱采用瑞士bruker公司avanceiii400mhz核磁共振仪测定,氘代氯仿或氘代dmso做溶剂。
本发明提供aβ斑块的显像方法。在本显像方法的第一步中,将可检测量的式(i)所示的化合物引入组织或患者中。化合物通常是诊断组合物的部分并且通过本领域技术人员公知的方法给药至组织或患者。如以可检测量的式(i)所示的化合物引入患者并且在足以使化合物与aβ斑块结合的时间之后,非创伤性地检测化合物。或将式(i)所示的化合物引入患者,经足量的时间以使化合物与aβ斑块结合,自患者取组织样品,并脱离患者检测组织中的化合物。或自患者取组织样品并且将式(i)所示的化合物引入该组织样品。在足以使该化合物结合至aβ斑块的时间之后,检测化合物。
可以通过整体的或局部的给药途径将式(i)所示的化合物给药至患者。例如,可以将化合物给药至患者以使其递送至全身。可选地,可以将化合物给药至关注的特定的器官或者组织。例如,为了诊断或追踪患者的ad的进程,需要定位和定量脑内的淀粉样斑块。
术语“患者”是指人类和其他动物。本领域技术人员也熟知如何确定足以使化合物与aβ斑块结合的时间。通过将可检测量的式(i)所示的化合物引入患者,然后在给药后的各时间处检测化合物,可以容易地测定所需的时间。
术语“结合”是指化合物和aβ斑块之间的化学相互作用。结合的实例包括共价键、离子键、亲水-亲水相互作用、疏水-疏水相互作用和络合物。
本发明所述的显像手段为光学显像。
实施例1
本实施例的一种荧光化合物(i)的合成,具体合成步骤如下:
(1)合成中间体1
将4.77g的2,3,3-三甲基吲哚(30mmol)与3.22ml碘甲烷(7.08g,50mmol)在60ml乙醇中搅拌均匀,密封耐压容器,80℃反应12h。反应完成后,冷至室温,析出固体,抽滤,得到析出固体7.7g,即得中间体1;
(2)合成中间体2
将17.5mldmf(16.54g,225mmol)与18ml二氯甲烷混合均匀,冰浴并氮气保护下,逐滴慢慢滴加15ml三氯氧磷(24.68g,160mmol),搅拌20-40min后,加入4.6ml环己酮(4.37g,45mmol),升温到80℃回流反应4h后,冷至室温,将反应液缓慢倒入冰水中,析出固体,抽滤,得到7.5g固体,即中间体2;
(3)合成中间体3
将7.5g中间体1(25mmol)和7g中间体2(40mmol)溶于140ml正丁醇与60ml甲苯混合溶剂中,升温到80-130℃反应4h后,除去溶剂,柱层析分离,得到2.5g中间体3;
(4)合成荧光化合物dcsc-1
将400mg中间体3(1.2mmol)和800μl丙二腈(12mmol)溶于32ml甲醇中,再加入320μl饱和的碳酸钾甲醇溶液,室温搅拌3h,抽滤,得到202mg析出固体,即得dcsc-1,产率44.9%。所得产物的核磁氢谱图和碳谱图分别如图2和图3所示。产物鉴定数据如下:
1hnmr(400mhz,dmso)δ8.00(d,j=13.8hz,1h),7.82(s,1h),7.48(d,j=6.9hz,1h),7.33(t,j=7.7hz,1h),7.20(d,j=7.6hz,1h),7.10(t,j=7.4hz,1h),5.90(d,j=12.9hz,1h),3.47(s,3h),2.81(t,j=8.7hz,2h),2.60(t,j=9.3hz,2h),1.85–1.73(m,2h),1.60(s,6h);
13cnmr(101mhz,cdcl3)δ166.28,153.40,148.14,143.74,139.64,137.59,128.17,124.81,123.96,122.35,121.94,117.37,115.59,107.87,95.15,72.11,47.32,29.82,29.71,28.34,27.49,25.86,20.96。
(5)合成荧光化合物dcsc-7
将400mg中间体3(1.2mmol)和1.28ml氰基乙酸乙酯(12mmol)溶于32ml甲醇中,再加入100μl哌啶,室温搅拌12h后,柱层析分离,即得126mg的dcsc-7,产率24.9%。所得产物的核磁氢谱图和碳谱图分别如图4和图5所示。产物鉴定数据如下:
1hnmr(400mhz,dmso)δ8.45(s,1h),7.86(d,j=14.1hz,1h),7.42(d,j=7.6hz,1h),7.28(t,j=8.2hz,1h),7.09(d,j=8.7hz,1h),7.02(t,j=7.1hz,1h),5.75(d,j=12.9hz,1h),4.25(q,j=14.5,7.5hz,2h),3.37(s,3h),2.88(t,j=5.8hz,2h),2.60(t,j=8.3hz,2h),1.79(m,2h),1.59(s,6h),1.26(t,j=7.5hz,3h);
13cnmr(101mhz,cdcl3)δ165.13,164.59,164.31,164.12,151.40,147.50,147.22,144.13,139.45,134.67,134.38,128.00,125.24,124.41,121.85,121.55,107.26,94.28,61.93,52.85,46.85,29.71,29.58,28.32,26.02,21.25,14.29。
实施例2
本实施例的一种荧光化合物(i)的合成,具体合成步骤如下:
(1)合成中间体1
将4.77g的2,3,3-三甲基吲哚(30mmol)与3.22ml碘甲烷(7.08g,50mmol)在60ml乙醇中搅拌均匀,密封耐压容器,80℃反应12h。反应完成后,冷至室温,析出固体,抽滤,得到析出固体7.7g,即得中间体1;
(2)合成中间体2
将17.5mldmf(16.54g,225mmol)与18ml二氯甲烷混合均匀,冰浴并氮气保护下,逐滴慢慢滴加15ml三氯氧磷(24.68g,160mmol),搅拌20-40min后,加入4.6ml环己酮(4.37g,45mmol),升温到80℃回流反应4h后,冷至室温,将反应液缓慢倒入冰水中,析出固体,抽滤,得到7.5g固体,即中间体2;
(3)合成中间体3
将7.5g中间体1(25mmol)和7g中间体2(40mmol)溶于140ml正丁醇与60ml甲苯混合溶剂中,升温到80-130℃反应4h后,除去溶剂,柱层析分离,得到2.5g中间体3;
(4)合成荧光化合物dcsc-1
将400mg中间体3(1.2mmol)和800μl丙二腈(6mmol)溶于16ml甲醇中,再加入160μl饱和的碳酸钾甲醇溶液,室温搅拌3h,抽滤,得到184mg析出固体,即得dcsc-1,产率40.9%。产物鉴定数据同实施例1。
(5)合成荧光化合物dcsc-7
将400mg中间体3(1.2mmol)和1.28ml氰基乙酸乙酯(6mmol)溶于16ml甲醇中,再加入50μl哌啶,室温搅拌12h后,柱层析分离,即得121mgdcsc-7,产率23.7%。产物鉴定数据同实施例1。
实施例3
本实施例的一种荧光化合物(i)的合成,具体合成步骤如下:
(1)合成中间体1
将4.77g的2,3,3-三甲基吲哚(30mmol)与3.22ml碘甲烷(7.08g,50mmol)在60ml乙醇中搅拌均匀,密封耐压容器,80℃反应12h。反应完成后,冷至室温,析出固体,抽滤,得到析出固体7.7g,即得中间体1;
(2)合成中间体2
将17.5mldmf(16.54g,225mmol)与18ml二氯甲烷混合均匀,冰浴并氮气保护下,逐滴慢慢滴加15ml三氯氧磷(24.68g,160mmol),搅拌20-40min后,加入4.6ml环己酮(4.37g,45mmol),升温到80℃回流反应4h后,冷至室温,将反应液缓慢倒入冰水中,析出固体,抽滤,得到7.5g固体,即中间体2;
(3)合成中间体3
将7.5g中间体1(25mmol)和7g中间体2(40mmol)溶于140ml正丁醇与60ml甲苯混合溶剂中,升温到80-130℃反应4h后,除去溶剂,柱层析分离,得到2.5g中间体3;
(4)合成荧光化合物dcsc-1
将400mg中间体3(1.2mmol)和800μl丙二腈(18mmol)溶于48ml甲醇中,再加入150μl饱和的碳酸钾甲醇溶液,室温搅拌3h,抽滤,得到196mg析出固体,即得dcsc-1,产率43.6%。产物鉴定数据同实施例1。
(5)合成荧光化合物dcsc-7
将400mg中间体3(1.2mmol)和1.28ml氰基乙酸乙酯(18mmol)溶于48ml甲醇中,再加入150μl哌啶,室温搅拌12h后,柱层析分离,即得115mgdcsc-7,产率22.7%。产物鉴定数据同实施例1。
本发明所得荧光化合物(i)应用性能测试:
(1)荧光化合物制备的探针与aβ1-42聚集体的体外结合实验:
实验方法:取本发明所得荧光化合物dcsc-1或dcsc-7溶于二甲亚砜中,配制成10mm储备液,用pbs溶液稀释成1μm的待测液(探针(i)。先测得探针(i)的激发和发射光谱性质。选用aβ1-42蛋白在37℃水浴中培育aβ聚集体,用于模拟人脑内的aβ蛋白聚集体。将探针(i)与aβ1-42聚集体(5μm)混合,应用荧光分光光度计进行荧光检测。所得探针(i)与aβ1-42聚集体混合前后的荧光光谱图见图6。由图6可以看出,本发明的荧光化合物(i)的近红外荧光性质良好,达到了近红外区。其中,荧光化合物dcsc-1与aβ聚集体结合后,最大发射波长为685nm左右。荧光化合物dcsc-1与aβ1-42聚集体混合后的荧光强度大幅增强。
(2)荧光化合物(i)探针应用于ad转基因小鼠活体成像:
实验步骤:将荧光化合物dcsc-1制备成溶液探针(4mg/kg,10%吐温80,20%dmso,70%pbs)由尾静脉注射入ad转基因小鼠(tg,app/ps1双转,10月龄)和正常小鼠(wt,c57bl6,10月龄)体内,在异氟烷的持续麻醉下分别在10min、30min、60min进行小鼠动物成像图像采集(caliperlifescience,ivisluminaxr,激发波长取630nm,发射波长取685nm),分析结果。采用livingimagingsoftware进行成像结果分析。所得成像实验结果如图7所示。结果显示,荧光化合物(i)可以透过血脑屏障,脑部的荧光信号在10分钟左右达到峰值。10分钟之后,ad转基因小鼠的脑部荧光信号强度要强于正常小鼠。荧光化合物dcsc-1在ad转基因小鼠体内的清除速率明显要慢于正常小鼠,表明探针与小鼠脑内的aβ斑块结合后滞留在小鼠脑部,使ad小鼠脑部的荧光信号明显高于正常小鼠,适用于活体的荧光成像。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其它的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!
- 化合物及使用其的有机电激发光组件的制作方法与工艺
- 一种烟草中倍半萜类化合物的测定方法与流程
- 对有机含磷化合物有毒气体具有荧光响应的一维有机半导体纳米线/带及其制备方法和应用与流程
- 一种用于调节白光LED的荧光化合物及其制备方法和应用与流程
- 联咔唑化合物、光固化性组合物、其固化物、用于塑料透镜的固化性组合物和塑料透镜的制作方法与工艺
- 联咔唑化合物、光固化性组合物、其固化物、用于塑料透镜的固化性组合物和塑料透镜的制作方法与工艺
- 一种基于π‑π堆积化合物的荧光材料及其制备方法与流程
- 一种有机电致荧光化合物及其在电致发光器件中的应用的制作方法与工艺
- 一种有机电致荧光化合物及其在电致发光器件中的应用的制作方法与工艺
- 一种有机荧光化合物及其应用的制作方法与工艺