一种高效稳定的Mn4+掺杂氟化物发光材料及其制备方法与流程
本发明涉及稀土发光材料和照明显示技术领域,尤其是涉及一种高效稳定的mn4+掺杂氟化物发光材料及其制备方法。
背景技术:
白光led作为一种新型的固态光源,与传统的白炽灯和荧光灯等光源相比,它具有环保、节能、高效、响应快等优点,被誉为继白炽灯、荧光灯和高压气体放电灯三大光源之后的第四代绿色光源。在led光源中,荧光粉的性能决定了led的发光效率、显色指数、色温及使用寿命等技术指标,因此,荧光粉在白光led中具有举足轻重的地位,受到广泛关注。目前最常用的方法是将蓝光led芯片(发光波长440-480nm)与黄光荧光粉(如yag:ce或tag:ce)相结合,黄光荧光粉吸收部分蓝光led芯片发出的蓝光后发射出黄光,并与未被吸收的蓝光混合形成白光。但是采用这种方式只能获得相关色温(correlatedcolortemperature,cct)大于4500k的冷白光器件,同时,其显色指数(colorrenderingindex,cri)也较低,通常小于80。其主要原因在于常用黄光荧光粉发射光谱中的红光成份不足,导致难以获得低色温、暖色调以及高显色指数的白光led器件,而这正是白光led能在室内获得应用的关键。要想实现这一目标,一个有效的办法就是在白光led器件中添加适当的红光荧光粉,增强器件的红光发射。
mn4+离子激活的氟化物材料具有窄的发射峰,半峰宽小,是目前红光发光材料的研究热点。专利us2009/7497973公开了mn4+激活的a2mf6(a为k,na,rb等;m为ti,si,sn,ge等)红光荧光粉;其中通过将原料溶解在高浓度氢氟酸中,然后加热挥发共结晶得到产物;专利wo2009/119486公开了将金属如si溶解在氢氟酸和高锰酸钾的溶液中,反应得到氟化物产物;专利cn102732249a公开报道了通过先制备含有金属m的氟化物的第一种溶液和含有a的第二种溶液或固体形式的a的化合物,将两者混合,经反应后生成沉淀,得到产物;这些方法制备的氟化物发光材料都具有发光效率高,热稳定性好的优点。但这些方法都没有克服氟化物发光材料易水解的缺点,在长期使用或潮湿环境下容易水解,是发光效率降低甚至失效。专利us2007/0125984a1报道了在氟化物表面包覆一层无机材料(如tio2、al2o3、sio2)保护膜,避免mn4+直接与水接触产生水解作用;专利cn106479485a同样公开报道在荧光粉颗粒表面包覆一层硅酸钾-羟甲基纤维素钠-聚乙二醇混合物,提高荧光粉的耐高温高湿性能;这些公开介绍的方法均能够有效提高荧光粉的抗湿能力,但另一方面,降低了材料的发光效率。
技术实现要素:
为了解决现有技术的不足,本发明的目的在于提供一种高效稳定的mn4+掺杂氟化物发光材料及其制备方法,以解决氟化物发光材料耐高温耐湿性能不足的缺点,同时能够有效的提高荧光粉的发光效率。该方法具有制备工艺简单、原料来源广泛以及氢氟酸消耗量小的特点,适于工业化大规模制备。
本发明的目的通过以下技术方案实现:
一种改性mn4+掺杂氟化物发光材料的制备方法,所述发光材料为表面被a2mf6包覆的mn4+掺杂氟化物a2mf6:mn4+;
其中,a选自碱金属li,na,k,rb,cs中的一种或它们之间的组合;
m选自ti,si,ge,sn,zr中的一种或它们之间的组合;
mn4+为发光中心离子;
其特征在于,所述方法包括如下步骤:
(1)将a2mf6溶于hf溶液中形成饱和溶液;
(2)将饱和溶液与a2mf6:mn4+发光材料混合,搅拌浸泡,经过滤干燥后,获得一次处理后的a2mf6:mn4+粉体;
(3)将步骤(1)的饱和溶液与步骤(2)过滤干燥后获得的a2mf6:mn4+粉体混合,再次搅拌浸泡,经过滤干燥后,获得二次处理后的a2mf6:mn4+粉体,重复上述步骤,制备得到所述改性的mn4+掺杂氟化物发光材料。
根据本发明,步骤(3)中,所述重复步骤的次数为1-6次,优选2-5次。
根据本发明,所述m优选为si和/或ti。
根据本发明,所述碱金属a优选为na和/或k。
根据本发明,所述mn4+掺杂氟化物a2mf6:mn4+选自k2sif6:mn4+、k2tif6:mn4+、k2snf6:mn4+、cs2sif6:mn4+、rb2gef6:mn4+、rb2sif6:mn4+、na2tif6:mn4+、na2sif6:mn4+、na2zrf6:mn4+、k2gef6:mn4+,优选为na2tif6:mn4+、na2sif6:mn4+、k2tif6:mn4+、k2sif6:mn4+。
根据本发明,所述a2mf6为相对应的基质化合物。
根据本发明,所述a2mf6选自k2sif6、k2tif6、k2snf6、cs2sif6、rb2gef6、rb2sif6、na2tif6、na2sif6、na2zrf6、k2gef6,优选为na2tif6、na2sif6、k2tif6、k2sif6。
根据本发明,步骤(1)中,所述氢氟酸的质量百分比优选20-50%。
根据本发明,步骤(1)中,所述饱和溶液优选在20℃-120℃下形成。
根据本发明,步骤(2)和(3)中,所述搅拌浸泡的温度为0-120℃,优选为20-50℃。
根据本发明,步骤(2)和(3)中,所述搅拌浸泡可以是在保温条件下进行,也可以在缓慢降温条件下进行,优选地,所述缓慢降温速率为0.1℃/min-10℃/min,还优选地0.5℃/min-5℃/min。
根据本发明,步骤(2)和(3)中,所述搅拌浸泡的时间为2min-2h,优选为5min-1h。
根据本发明,步骤(2)和(3)中,所述a2mf6和a2mf6:mn4+的摩尔比为1-5:1。
根据本发明,步骤(2)和(3)中,经过滤后的产物可进一步用有机溶剂如无水乙醇或丙酮进行洗涤,以去除表面残余的酸液,并进行干燥。
本发明中,经所述方法会使a2mf6:mn4+表面的mn4+与饱和a2mf6溶液中的m4+实现离子交换,除去了a2mf6:mn4+表面的mn4+,在荧光粉表面形成a2mf6化合物保护层,同时减少激活离子mn4+在a2mf6:mn4+荧光粉表面形成的缺陷。
本发明中,当所述搅拌浸泡过程在保温条件下进行时,经过滤干燥后的粉末样品,表面层为a2mf6化合物的a2mf6:mn4+氟化物荧光粉,荧光粉颗粒尺寸没有变化,该过程中,a2mf6:mn4+氟化物荧光粉表层的mn4+和溶液中的m离子发生离子交换反应,因此,该荧光粉颗粒的尺寸没有变化。
本发明中,当所述搅拌浸泡过程在缓慢降温条件下进行时,经过滤干燥后的粉末样品,表面层为a2mf6化合物的a2mf6:mn4+氟化物荧光粉,荧光粉颗粒尺寸增大,该过程中,a2mf6:mn4+氟化物荧光粉主要发生析晶包覆,即溶液中的a2mf6化合物随着温度的变化,析出并包覆在a2mf6:mn4+氟化物荧光粉表层,因此,该荧光粉颗粒尺寸增大。
本发明还提供一种改性mn4+掺杂氟化物发光材料,其是采用上述方法制备得到的。
本发明的有益效果:
本发明提供了一种高效稳定的改性mn4+掺杂氟化物发光材料及其制备方法,所述制备方法能在0-120℃的温度范围内高效的合成a2mf6化合物包覆的a2mf6:mn4+氟化物荧光粉,避免mn4+直接与外界环境接触发生降解,具有高温稳定性和良好的抗湿性能,并且进一步提高了荧光粉的发光效率。该方法制备温度低、时间短、工艺易控制,适合工业大规模制备。
附图说明
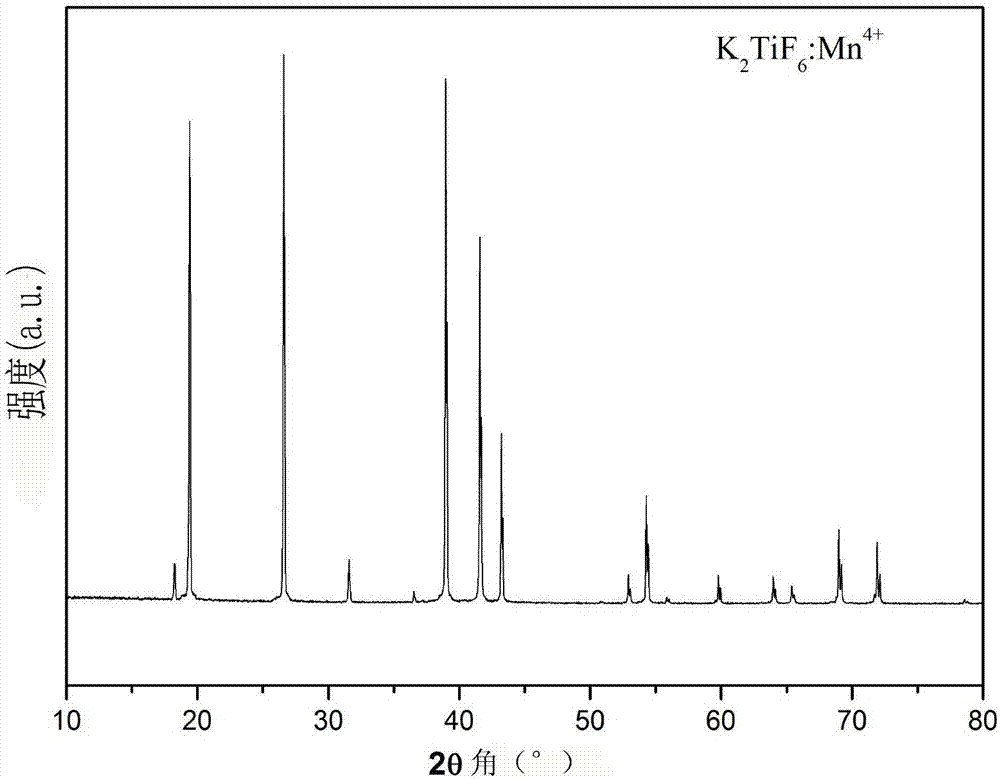
图1本发明实施例2中k2tif6:mn4+荧光粉的xrd衍射图谱。
图2本发明实施例2中k2tif6:mn4+荧光粉的激发光谱和发射光谱。
图3本发明实施例6中k2sif6:mn4+荧光粉的xrd衍射图谱。
图4本发明实施例6中k2sif6:mn4+荧光粉的激发光谱和发射光谱。
具体实施方案
下面结合实施例对本发明进行进一步的说明。但本领域技术人员了解,下述实施例不是对本发明保护范围的限制,任何在本发明基础上做出的改进和变化,都在本发明的保护范围之内。
实施例1:k2tif6:mn4+荧光粉的制备
将0.4000gk2mnf6溶解在6ml氢氟酸(49wt.%)溶液中,搅拌5分钟得到橙黄色透明溶液,然后将5.1850gk2tif6粉末加入到溶液中,室温下继续搅拌30分钟,停止搅拌并用滤纸过滤,再用丙酮进行清洗,出去任何残留的hf,经过干燥,得到k2tif6:mn4+粉末。通过fls980型(edinburghinstrument)荧光光谱仪测量了产物的激发和发射光谱以及荧光量子产率和吸收效率。
实施例2:改性的k2tif6:mn4+荧光粉的制备
通过向10ml49%hf溶液中加入k2tif6化合物,直至不再溶解为止(约1.7000g,室温下),过滤未溶解的k2tif6,获得k2tif6在49%hf溶液中的饱和溶液。随后将该饱和溶液加入到装有实施例1中获得的k2tif6:mn4+荧光粉的容器中,在室温下持续搅拌30min。随后进行真空抽滤;重复上述搅拌浸泡步骤2次,最后过滤后用丙酮洗涤数次,除去残留hf,经过70℃干燥2h后,获得荧光粉体。x射线粉末衍射表明产物仍为六方相k2tif6结构(图1);通过fls980型(edinburghinstrument)荧光光谱仪测量了产物的激发和发射光谱以及内荧光量子产率,图2为其激发和发射光谱,表1给出了所制备荧光粉的重要物化和光学性能参数,包括mn4+的掺杂浓度,制备原料配比以及样品的荧光量子产率和吸收效率。
实施例3:改性的k2tif6:mn4+荧光粉的制备
通过向10ml49%hf溶液中加入k2tif6化合物,溶液加热至70℃并不断搅拌直至不再溶解为止(约2.3000g),过滤未溶解的k2tif6,获得k2tif6在70℃下49%hf溶液中的饱和溶液。随后将该饱和溶液加入到装有实施例1中获得的k2tif6:mn4+荧光粉的容器中,以0.5℃/min的降温速度缓慢降温,并持续搅拌至室温。随后进行真空抽滤,用丙酮洗涤数次,除去残留hf,经过70℃干燥2h后,获得荧光粉体。表1给出了所制备荧光粉的重要物化和光学性能参数,包括mn4+的掺杂浓度,制备原料配比以及样品的荧光量子产率和吸收效率。
实施例4:稳定性测试
分别将实施例1,实施例2和实施例3制备获得的荧光粉样品在水中浸泡一定时间,测试其发光强度值对比。分别取实施例1,实施例2和实施例3荧光粉样品0.5000g,量取1ml蒸馏水加入样品中,分别浸泡10min,经过滤干燥后,分别测的水中浸泡后发光强度如表1所示。
表1k2tif6:mn4+荧光粉处理前后荧光性能对比
由表中可以看出,改性处理后提高了荧光粉的量子产率,更重要的是提高了荧光粉的抗湿稳定性,在水中浸泡后发光强度衰减少。
实施例5:k2sif6:mn4+荧光粉的制备
将0.3250gk2mnf6溶解在4ml氢氟酸(49%)溶液中,搅拌5分钟得到橙黄色透明溶液,然后将5.5000gk2sif6粉末加入到溶液中,并在室温下继续搅拌3小时,停止搅拌后用滤纸过滤得到k2sif6:mn4+粉末,x射线粉末衍射表明产物具有立方相k2sif6结构,在蓝光激发下,该荧光粉发出明亮的红光。
实施例6:改性的k2sif6:mn4+荧光粉的制备
通过向10ml49%hf溶液中加入k2sif6化合物,直至不再溶解为止(约0.1750g,室温下),过滤未溶解的k2sif6,获得k2sif6在49%hf溶液中的饱和溶液。随后将实施例5中获得的k2sif6:mn4+荧光粉加入到该饱和溶液中,在室温下持续搅拌30min。随后进行真空抽滤,重复上述搅拌浸泡步骤2次,最后过滤后用丙酮洗涤数次,除去残留hf,经过70℃干燥2h后,获得荧光粉体。x射线粉末衍射表明产物仍为六方相k2sif6结构(图3);通过fls980型(edinburghinstrument)荧光光谱仪测量了产物的激发和发射光谱以及内荧光量子产率,图4为其激发和发射光谱,表2给出了所制备荧光粉的重要物化和光学性能参数,包括mn4+的掺杂浓度,制备原料配比以及样品的荧光量子产率和吸收效率。
实施例7:改性的k2sif6:mn4+荧光粉的制备
通过向10ml49%hf溶液中加入k2sif6化合物,溶液加热至70℃并不断搅拌直至不再溶解为止(约0.2200g),过滤未溶解的k2sif6,获得k2sif6在70℃下49%hf溶液中的饱和溶液。随后将该饱和溶液加入到装有实施例5中获得的k2sif6:mn4+荧光粉的容器中,以0.5℃/min的降温速度缓慢降温,并持续搅拌至室温。随后进行真空抽滤,用丙酮洗涤数次,除去残留hf,经过70℃干燥2h后,获得荧光粉体。表2给出了所制备荧光粉的重要物化和光学性能参数,包括mn4+的掺杂浓度,制备原料配比以及样品的荧光量子产率和吸收效率。
实施例8:稳定性测试
分别将实施例5,实施例6和实施例7制备获得的荧光粉样品在水中浸泡一定时间,测试其发光强度值对比。分别取实施例5,实施例6和实施例7荧光粉样品0.5000g,量取1ml蒸馏水加入样品中,分别浸泡10min,经过滤干燥后,分别测的水中浸泡后发光强度如表2所示。
表2k2sif6:mn4+荧光粉处理前后荧光性能对比
由表中可以看出,改性处理后提高了荧光粉的量子产率,更重要的是提高了荧光粉的抗湿稳定性,在水中浸泡后发光强度衰减少。
以上,对本发明的实施方式进行了说明。但是,本发明不限定于上述实施方式。凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!