制备纯三甲硅烷基胺的方法与流程
在本发明的上下文中,将三甲硅烷基胺缩写为TSA,二甲硅烷基胺缩写为DSA,一氯硅烷缩写为MCS。
已知将TSA用于生成氮化硅层,如文献US 4,200,666和JP 1986 96741中所述。特别是将TSA作为氮化硅或氮氧化硅层的层前体用于芯片制备中,如US 2011/0136347中所述。以WO 2004/030071公布的专利申请公开了一种应用TSA的具体工艺,其中明确说明,以稳定的高品质安全无失误地制备TSA对于其在芯片制备中的应用尤其重要。
TSA的制备依照以下反应方程式进行:
(1)3SIH3Cl+4NH3→3NH4Cl+(SiH3)3N。
该反应由Stock和Somieski于1921年首次提出[1]。当时该反应以气相进行。
此文献中,对于从氨和MCS合成TSA描述了两种反应机理。
在文献[2]中,按照以下三步反应描述了一种反应机理。
(2)
(3)
(4)
这种情形中,MCS与氨在第一个反应式中反应生成加合物,该加合物与另外的氨分子反应完全以生成单甲硅烷基胺和氯化铵。
在第二反应式中,单甲硅烷基胺与另外的MCS分子反应生成加合物,该加合物与另外的氨分子反应完全以生成二甲硅烷基胺和氯化铵。
在第三反应式中,二甲硅烷基胺与另外的MCS分子反应生成加合物,该加合物与另外的氨分子反应完全最终形成三甲硅烷基胺和氯化铵。
根据文献[2,3],关于反应机理,还另外将缩合反应(6)和(7)纳入考虑。
(5)
(6)6SiH3NH2→3(SiH3)2NH+3NH3
(7)3(SiH3)2NH→2(SiH3)3N+NH3
这种情形中,MCS与氨在第一反应式中反应生成加合物,该加合物与另外的氨分子反应完全以生成单甲硅烷基胺和氯化铵。
在第二反应式中,单甲硅烷基胺缩合或歧化,生成二甲硅烷基胺和氨。
在第三反应式中,二甲硅烷基胺缩合或者歧化,生成三甲硅烷基胺和氨。
不仅依据反应式(2)、(3)和(4)的反应机理,还有依据反应式(5)、(6)和(7)的反应机理,均是以单甲硅烷基胺[12]和二甲硅烷基胺(参见[6]、[7]、[8]和[12]以及本说明书)为中间产物的三级反应机理。
Miller[6]描述了一种制备TSA的设备和方法。这种情形中,MCS和氨以气态流过反应器。流出反应器的气体混合物在下游-78℃的冷阱中凝出。流出反应器的气体混合物和/或冷凝液含有甲硅烷、MCS、DSA和TSA。将冷阱加热到20℃后,液体含有甲硅烷、MCS和TSA。
Ritter[7]描述了用苯甲醚作溶剂的TSA液相合成工艺。将MCS注入苯甲醚中,向该溶液中添加氨。
Aylett和Hakim[4]公开了一种工艺,当进行该工艺时,在将气相加热到150℃3个小时后,DSA保持不变。此外,他们还报道,在0℃下72小时后,液相中80%的DSA依据以下反应式转化成TSA。
(8)3(SiH3)2NH→2(SiH3)3N+NH3。
此外,还报道,DSA与过量的氨在室温下不以气相反应,在-130℃下在1分钟内DSA全部分解生成硅烷和少量氨。
Wells和Schaeffer[5]描述了MCS和氨在反应杯(reaction cuvette)中从-196℃加热到室温下的缩合反应。在这种情形中,除了TSA、甲硅烷和氨,还形成了聚硅氮烷和氯化铵。
Korolev[8]描述了用甲苯作溶剂的TSA液相合成工艺。将MCS注入甲苯,向该溶液中添加氨。将该混合物在约-100℃至0℃的温度下搅拌约1-48小时。并不清楚该时间要求仅与添加氨的时长相关,还是无论是否如此,可能非排他地表示仅与添加完成之后进行搅拌的时长相关。示例性实施方案表明反应后将混合物在室温下搅拌24小时。由此可以得出结论,进行该方法的必要时长大于24小时。
文献[6]和[7]中称,氨的卤化物如氯化铵是催化剂,在其存在下,TSA歧化成硅烷或者其他分解产物。结果,TSA收率下降。
在文献[7]的所有示例性实施方案中,除了实施例9和10,均采用明显过量的MCS。以过量的MCS操作意味着MCS通过蒸馏进入后处理,并且作为DSA与MCS的反应结果在此产生氯化铵沉积物,生成氯化铵。
实施例9记载MCS缺乏26mol%。如果如同表1的实施例1-7,实施例9中所列TSA收率85%是基于氨,那么通过计算会得到TSA基于MCS的不可能的收率115%。
实施例10中公开了添加两倍化学计量量的氨。结果表明没有形成TSA。仅检测到甲硅烷和氨。
除了第11个实施例中TSA的收率(68%)之外,文献[7]在示例性实施方案1-8和11-13中披露TSA基于MCS的收率在14%~58%之间。其原因主要在于MCS相对于氨的极大过量。
文献[8]实施例1中披露,化学计量过量的MCS致使MCS通过蒸馏进入后处理,并且作为DSA与MCS的反应结果在此产生氯化铵沉积物。
文献[8]示例性实施方案中,(实施例1)以化学计量不足的NH3操作,TSA基于MCS的收率是57%;(实施例2)以化学计量比的NH3∶MCS操作,收率为63%;实施例3中以化学计量过量的NH3操作,收率为34%。这说明化学计量过量的氨导致TSA收率降低,并且生成“不期望的”副产物。因此,MCS与氨的化学计量摩尔比优选1∶1至1.5∶1。另外,这也说明过量的MCS生成良好收率和品质的TSA。因此,MCS与氨的化学计量摩尔比特别优选1.1∶1至1.5∶1(参见文献[8]的[0045]段)。
对于使用过量NH3操作模式的情形,文献[8]的实施例3并未表明除了TSA之外还生成DSA。而且,还观察到由氨与TSA之间的缩合反应生成的产物。
文献[8]中表明,经蒸馏纯化的TSA具有约97%mol/mol至约100%mol/mol的纯度。根据示例性实施方案,TSA的纯度为91%mol/mol(实施例1)、92%mol/mol(实施例2)和40%mol/mol(实施例3)。
文献[7]中未提及所得TSA的纯度。
文献[9,10,11]中记载了采用惰性溶剂优选甲苯在液相中合成TSA。
专利申请[10]公开了一种耦合生产(coupled production)聚硅氮烷和三甲硅烷基胺的工艺,其中TSA和聚硅氮烷通过一氯硅烷与添加的初始化学计量量的氨进行反应来制备。TSA随后以气态形式从产物混合物中分离。紧接着补加氨,由此在此步骤中第一次得到相对于一氯硅烷的初始注入量所引入的总氨量化学计量过量。由于向反应器添加初始化学计量量的氨,一氯硅烷反应不完全。从而,在随后的气态TSA分离中,一氯硅烷和少量生成的二甲硅烷基胺也进入TSA产物溶液。二甲硅烷基胺和一氯硅烷相互反应。该反应缓慢进行并伴有更多的氯化铵的沉积。结果,氯化铵的沉积在从反应器移出的TSA产物溶液中或者在反应器下游设备部件中产生。由于该缓慢反应,氯化铵的沉积在过滤之后的TSA产物滤出液中再次发生。特别是,该反应导致氯化铵沉淀在用于纯化TSA的精馏塔中。
专利申请[11]公开了一种从一氯硅烷和氨耦合生产聚硅氮烷和三甲硅烷基胺的工艺,完全避免了文献[10]中所述的缺点和限制,特别是随后在反应器外纯化TSA产物流的制备部件中防止一氯硅烷与二甲硅烷基胺反应生成氯化铵。
为此目的,直接在一个步骤中添加相对于存在于惰性溶剂中的一氯硅烷化学计量过量的氨。由于以相对于一氯硅烷化学计量过量的量引入NH3,一氯硅烷在反应器中完全反应。由此,通过引入相对于一氯硅烷化学计量过量的NH3,防止了设备的下游部件中一氯硅烷与少量形成的其它二甲硅烷基胺反应生成固体氯化铵。
含有TSA的产物混合物随后以气态形式分离。将所得产物混合物过滤然后完全脱除氯化铵。经精馏纯化并由此得到高纯度或者超高纯度的TSA。精馏后所用精馏塔不含任何固体氯化铵。
人们对于以相对较高纯度提供TSA的商业工艺始终存在巨大的兴趣。
本发明的目的是提供一种方法,其尽可能完全地合成TSA且不生成显著量DSA。该目的包括尽可能地避免现有技术中所见的TSA经氯化铵催化的分解反应生成硅烷和其他分解产物。
本发明提供一种方法,该方法实施过程中,根据反应式(2)、(3)和(4)或(5)、(6)和(7)的三步反应进行完全,在产物中不剩余显著量的DSA。这相当于,将氨添加到反应器后,各反应步骤迅速进行,生成TSA,避免了氨的不完全硅烷化反应以及由此而来的仅导致生成DSA的反应进程,除了少量残余量的DSA之外。
本发明人的观点认为,试剂一氯硅烷和氨的浓度、反应器的温度以及剧烈搅拌对于三步TSA反应的速度和完成程度都有决定性影响。
本发明涉及一种液相制备三甲硅烷基胺的方法,其中:
(a)至少将溶解于溶剂(L)中的一氯硅烷(MCS)以液态注入反应器(1)中,其中:
所述溶剂相对于MCS、氨(NH3)和TSA呈惰性,具有比TSA高的沸点;搅拌溶液,并
将溶液的温度T设定为10℃或更高;以及
(b)在反应器(1)中进行反应,其中:
将相对于MCS化学计量过量的NH3引入反应器(1)中,
其中保持温度T,然后
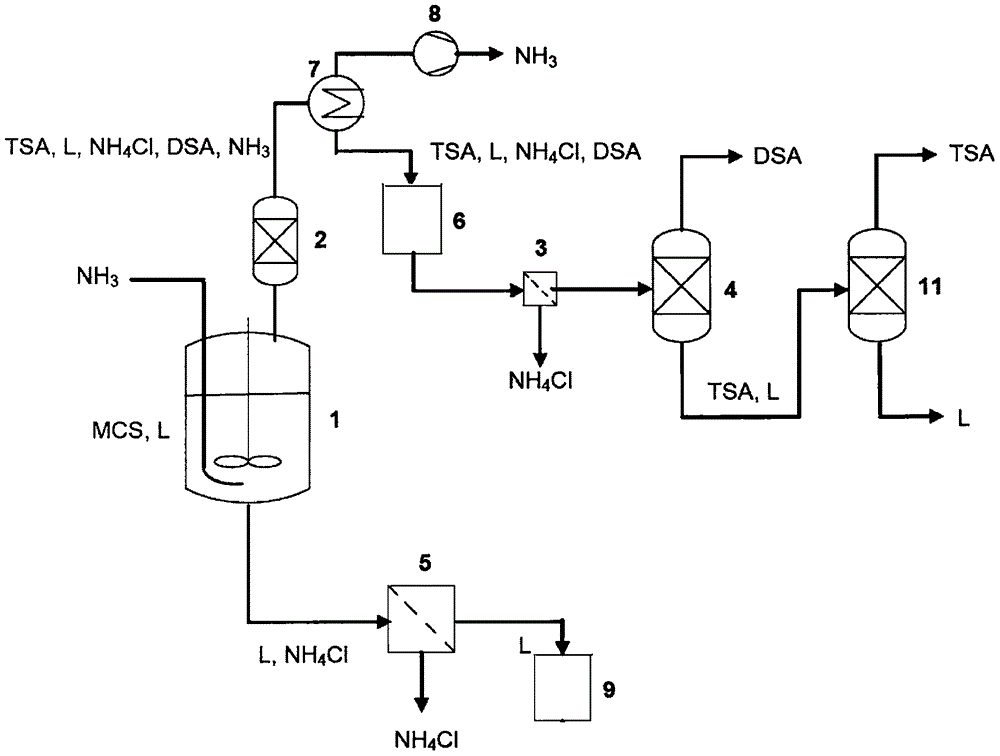
(c)将反应器减压,设定压力从0.5巴绝对压力(bar a)到0.8巴绝对压力;加热反应器,引导产物混合物(TSA、L、NH4Cl、DSA、NH3)以气态形式流出反应器(1)塔顶、流经蒸馏单元(2),经真空单元(8)分离NH3,在换热器(7)中冷凝产物混合物(TSA、L、NH4Cl、DSA),在容器(6)中收集产物混合物(TSA、L、NH4Cl、DSA);然后
(d)用过滤单元(3)过滤产物混合物,从产物混合物分离固体氯化铵(NH4Cl),将滤出液从过滤单元(3)导入精馏塔(4),
其中在塔顶分离DSA与混合物(TSA、L),在塔顶分离出DSA,将混合物(TSA、L)引入精馏塔(11),
其中在塔顶分离TSA与溶剂(L),在塔顶分离出TSA,并将溶剂再循环,或者
将滤出液从过滤单元(3)引入间歇式精馏塔(4),由此,先从塔顶分离出DSA,然后从塔顶分离出TSA,并将溶剂再循环,
以及
(e)引导底部产物混合物(L、NH4Cl)从反应器(1)流过过滤单元(5),在其中分离固体氯化铵(NH4Cl),得到溶剂(L)并收集在容器(9)中,然后
(f)将0-99%的将溶剂再循环,未再循环的溶剂由溶剂(L)替代。
该方法具有反应速率高的优点,该优点由选择高于0℃的温度、通过搅拌对溶液中的试剂进行剧烈混合以及通过使用液态MCS而具有的高浓度MCS而获得。由此,TSA的合成以高生成速率进行,也即DSA向TSA快速转化。该方法由此获得高的空时收率。
本发明的方法的另一个优点在于,完成步骤b之后甚至很短的时间就可以从反应器蒸馏分离TSA-溶剂混合物,因为,完成步骤b时,TSA的形成已经实质上进行完全了。有利地,在完成步骤b中添加NH3之后的至多12小时、优选8小时的间隔内,蒸馏出的至少部分量的TSA已经存在。由此,TSA在反应器中的停留时间相当短。不必再等待现有技术[8]中所知添加氨之后为TSA可得且随之可被纯化和分离所需的时间。
通过本发明的方法,TSA的收率提高。本发明人推测,反应器中TSA的短停留时间和接触时间有助于这个有利的效果,因为这减少了反应器中不期望的歧化反应或TSA与过量氨的其他反应。
此外,本发明方法的优点还在于,在步骤d和e中都得到溶剂(L),如果该方法以一次以上间歇进行,原材料可以节省地加入到步骤a所用的溶剂L中。
步骤d之后所得TSA具有至少99.5重量%的纯度。本发明所用相对于MCS化学计量过量的NH3具有MCS在反应器中完全反应的优点。这就防止了MCS通过蒸馏进入后处理并在此与DSA反应生成氯化铵。所生成的氯化铵会形成沉积物,不利于蒸馏后处理中的处理。
该方法获得高的和/或具有技术经济利益的TSA基于MCS的收率。特别地,在本发明的操作模式中,获得相对于现有技术提高的TSA收率和高于99.5重量%的TSA纯度。因此,本发明方法也具有所生成的TSA适用于半导体工业中的加工处理的优点。
以下详细阐述本发明的方法。
步骤b中需要监测温度T。因为反应是放热的,必须采用本领域技术人员已知的方法散去反应焓,保持温度。
优选地,在该方法的步骤b中,可以采用使NH3化学计量过量0.5~20%的氨量,相应地,MCS∶NH3的化学计量摩尔比为0.995~0.833。
优选地,在该方法的步骤b中,可以采用使NH3化学计量过量0.5~10%的氨量,相应地,MCS∶NH3的化学计量摩尔比为0.995~0.909。
特别优选地,在该方法的步骤b中,可以采用使NH3化学计量过量0.5~5%的氨量,相应地,MCS∶NH3的化学计量摩尔比为0.995~0.953。
步骤c中,以本领域技术人员已知的方式加热反应器,以通过蒸馏从反应器中的悬浮液分离产物混合物。蒸馏开始时未反应的过量NH3逸出,随后DSA分出,然后是TSA,之后是溶剂。持续蒸馏直到最后仅有纯溶剂分出。利用该方法的步骤c和d,NH3与TSA的二级反应被抑制,因为在完成NH3添加后的短暂时段之后,产物混合物就开始从反应器蒸馏分离。在这种情形下,氨实质上经真空泵完全流入废气中。仍然存在于所收集的含有TSA、DSA和溶剂的冷凝物中的极低残余量的氨在后续精馏处理中与DSA一并除去,所述精馏处理用于从相应精馏塔的塔顶分离除去DSA。
步骤d中所得溶剂可以全部再循环。这不仅适用于精馏塔的间歇操作模式,也适用于连续操作模式。
有利的是,在步骤f中再循环所回收溶剂的0-99%,并且用新鲜溶剂(L)替代未再循环的溶剂。
优选地,采用不会与TSA或DSA形成共沸物的惰性溶剂。优选该惰性溶剂应当比TSA/不易挥发和/或沸点比三甲硅烷基胺高至少10K。这类优选的溶剂可以选自烃、卤代烃、卤碳、醚、聚醚、叔胺。
特别优选采用甲苯作为溶剂(L)。这种选择的优点是TSA在甲苯中稳定。此外,氯化铵在甲苯中有限溶解,这有助于通过过滤除去氯化铵。
为获得高反应速率所需的高试剂浓度通过采用经溶剂(L)稀释的液相一氯硅烷来获得。本发明方法中有利的是采用对MCS体积过量的溶剂(L),优选甲苯。优选地,设定液体溶剂对MCS的体积比为30∶1-1∶1,优选20∶1-3∶1。特别优选地,步骤a中MCS可用溶剂以溶剂∶MCS体积比为10∶1-3∶1来稀释。然而,以3∶1-1∶1的体积比范围则优势变小。体积过量的溶剂确保了MCS的稀释。这也促成反应期间反应溶液中形成氯化铵的浓度降低的优点,由此有利于反应器的搅拌和排空。此外,文献[6]和[7]中所述由氯化铵催化的TSA的分解也减少。然而,量L的过大体积过量如高于30∶1,则降低反应器内的空时收率。
确实已知,温度的影响、通常提高10K会使反应速率加倍。然而,文献[8]中,制备TSA的反应在-100℃-0℃的温度下进行。这是因为担心在较高的温度下因更利于形成聚硅氮烷而降低TSA收率。假定在此温度下,反应式(2)-(5)中间所示加合物热力学不稳定,易于分解生成不期望的聚硅氮烷,由此TSA收率降低。
相反,本发明方法中令人惊异地发现,在10℃或更高的温度下,仅以难以察觉的微量生成聚硅氮烷。
因此在该方法的步骤a中优选将温度设定为10℃-30℃并在步骤b中保持,特别优选10℃-20℃,尤其优选设定为10℃的温度。
对于反应器的剧烈彻底混合,可以采用搅拌器以同时获得两种益处。第一,计量供给到反应器的氨可被直接分散以避免氨的局部高浓度,使引入的氨可以精细分散,从而抑制生成聚硅氮烷的副反应。第二,通过搅拌,反应器中形成的氯化铵可以悬浮且保持在悬浮液中以避免沉积。搅拌器的选择是本领域技术人员已知的。
提供一种方法,其中TSA合成伴以氨的计量供给以准原位进行,该方法将反应器内的温度设定为10℃或更高,优选10℃-30℃、更优选10℃-20℃、特别优选10℃,液体溶剂对一氯硅烷的体积比为30∶1-1∶1、优选20∶1-3∶1,更优选10∶1-3∶1,并采用配有搅拌器搅拌的高压釜,在该高压釜内直接分散计量供给的氨,使生成的氯化铵悬浮并保持在悬浮液中。相应地,氨的计量可以在宽范围内变动,可以提高以获得对技术操作有利的空时收率。同时,由于TSA的准原位形成,定量供给氨之后所需的后反应时间减少,也即最多需要1小时的必要后搅拌时长。后搅拌在步骤a和b的温度下进行。现有技术中,特别是根据Korolev的文献[8]中,需要长达48h的显著更长的后搅拌时间。
相反地,在本发明方法中,在计量供给NH3结束之后最多1h,在步骤c中将反应器减压,设定0.5巴绝对压力-0.8巴绝对压力的蒸馏压力,将搅拌高压釜加热以进行之后的蒸馏,随后TSA同绝大部分甲苯一起从反应器蒸出。蒸出的溶液进料给精馏步骤以制备纯TSA。
进行加热以将TSA与DSA、NH3、与部分L以及少量NH4Cl蒸馏出反应器。为此目的,引导产物混合物(TSA、L、NH4C1、DSA、NH3)以气态形式流出反应器(1)塔顶、流经蒸馏单元(2),利用真空单元(8)分离NH3,在换热器(7)中冷凝产物混合物(TSA、L、NH4Cl、DSA),在容器(6)中收集产物混合物(TSA、L、NH4Cl、DSA)。
在蒸馏过程中,NH3首先逸出,穿过换热器(7)和真空单元(8)进入废气。随后,在换热器(7)中保持DSA在设定压力下的冷凝温度一小段时间,例如0.5巴绝对压力下约12℃。随后,在换热器(7)中保持TSA在设定压力下的冷凝温度,例如0.5巴绝对压力下约27℃。
纯TSA蒸馏期间保持冷凝温度恒定。一旦甲苯开始共蒸馏就立即升高换热器(7)中的冷凝温度。蒸汽中的甲苯馏分继续上升直到蒸出纯甲苯。此时,在换热器(7)中保持纯甲苯在设定压力下的冷凝温度,例如0.5巴绝对压力下约85℃。在蒸出足够量纯甲苯后,也即确保留在反应器内的底部混合物基本上不含TSA和DSA之后,停止加热反应器以终止蒸馏。
本领域技术人员已知,为开始蒸馏并获得第一滴TSA蒸馏物,减压和加热反应器所用时间随反应器体积而增加。在小体积的反应器中进行反应是有利的,优选1-10l,特别优选5l的的体积。优选地,一旦引入NH3完成后2小时,或者优选一旦在步骤a和b中在所保持温度下进一步搅拌完成后1小时,就能收集到第一滴TSA蒸馏物。
从引入NH3完成到第一滴TSA蒸馏物冷凝之间的时长,或者从步骤a和步骤b中在所保持温度下进一步搅拌完成到第一滴TSA蒸馏物冷凝之间的时长,取决于将反应器减压和加热所需的时间。本发明的方法允许在步骤a和b所保持温度下进一步搅拌1小时后直接开始蒸馏。
综上,所述方法确保提供TSA的高时空收率,特别是在技术经济方面。
本发明还涉及一种用于使溶剂(L)中的至少一氯硅烷(MCS)试剂与氨在液相中反应以制备三甲硅烷基胺(TSA)的设备,其包括:
-反应器(1),其具有组分氨、至少MCS和(L)的进料管线,以及产物混合物(TSA、L、NH4Cl、DSA、NH3)的出口,该出口通入
-反应器(1)下游的蒸馏单元(2)、连接真空泵(8)和容器(6)的换热器(7),容器(6)配有管线连接至
-过滤单元(3),其具有至少一个NH4Cl固体出口和传送滤出液的较远管线,该较远管线通入
-精馏塔(4),其配有塔顶DSA出口和混合物(TSA、L)从底部排出的排出设施,该排出设施通入
-精馏塔(11),其配有塔顶TSA出口和溶剂(L)从底部排出的排出设施,
或者通入
-间歇精馏塔,向其引入来自过滤单元(3)的滤出液,
以及底部产物混合物(L、NH4Cl)从反应器底部排出的排出设施,该设施通入
-下游过滤单元(5),具有至少一个NH4Cl固体出口和传送含有溶剂的滤出液的较远管线,该较远管线通入
-容器(9)。
对于连续顺序操作的精馏塔,或者间歇精馏塔,DSA首先从塔顶移出,然后TSA从塔顶移出。这两种情形中溶剂均可再循环。
从容器(9),0-99%的溶剂可再循环。未再循环的溶剂必须由溶剂(L)替代。实施这些操作所需的设备构件是本领域技术人员已知的。
以下参照实施例举例说明本发明的方法。
对比实施例1
将3400ml甲苯和469g一氯硅烷先后装入已经提前用惰性气体吹扫过的5l搅拌式高压釜中,该高压釜具有冷却和加热模式并且与蒸馏单元连接,蒸馏单元包含蒸馏塔和冷凝器。以7小时10分钟的时长向反应溶液中添加178g氨。添加期间,温度恒定在0℃。添加期间的压力保持在3巴绝对压力。
添加氨之后,将混合物在0℃下进一步搅拌1小时。然后将反应器溶液调节至-20℃并保持,继续进一步搅拌整夜。
第二天,通过蒸馏单元下游连接的真空泵将压力设定为0.5巴绝对压力并将搅拌式高压釜加热。通过蒸馏单元,将TSA、DSA、甲苯馏分和痕量氯化铵蒸馏分出。合成过程的过量氨经真空泵流入蒸馏的废气。蒸馏液冷凝器的低温恒温器在-20℃的流动温度下操作。完成上述向反应溶液的添加氨之后17小时40分钟,收集到第一滴TSA蒸馏物。收集到第一滴TSA蒸馏物之后1小时30分钟停止蒸馏。
将收集到的蒸馏溶液过滤,然后除去氯化铵即变得澄清。随后,通过精馏首先分离DSA出(7g)。然后通过精馏从甲苯(384g)分离TSA(172g)。
完成精馏操作之后,所用精馏塔既不含有固体也不含有沉积物。完成蒸馏之后,精馏塔下游的冷阱含有1.5g物质,经定性分析,该物质含有Si和N。
基于所用一氯硅烷,蒸馏分离的TSA的收率为68%。所得TSA的纯度高于99.5重量%。
将仍然位于搅拌式高压釜中的甲苯、氯化铵、少量TSA、DSA和聚硅氮烷的溶液排出并过滤。经过滤的甲苯含有6g TSA、0.5g DSA、3g聚硅氮烷,不含氯化铵。经干燥的氯化铵滤饼含有3g硅。
对比实施例2
将3400ml甲苯和470g一氯硅烷先后装入已经提前用惰性气体吹扫过的51搅拌式高压釜中,该高压釜具有冷却和加热模式并且与蒸馏单元连接,蒸馏单元包含蒸馏塔和冷凝器。以7小时10分钟的时长向反应溶液中添加179g氨。添加期间,温度恒定在0℃。添加期间的压力从2.6巴绝对压力升至2.8巴绝对压力。
添加氨之后,将混合物在0℃下进一步搅拌1小时。
然后通过蒸馏单元下游连接的真空泵将压力设定为0.5巴绝对压力并将搅拌式高压釜加热。通过蒸馏单元,将TSA、DSA、甲苯馏分和痕量氯化铵蒸馏分出;合成过程的过量氨经真空泵流入蒸馏的废气。蒸馏液冷凝器的低温恒温器在-20℃的流动温度下操作。完成上述向反应溶液添加氨之后2小时,收集到第一滴TSA蒸馏物。收集到第一滴TSA蒸馏物之后2小时10分钟停止蒸馏。
将收集到的蒸馏溶液过滤,然后除去氯化铵即变得澄清。随后,通过精馏首先分离出DSA(4g)。然后通过精馏从甲苯(319g)分离出TSA(173g)。
完成精馏操作之后,所用精馏塔不含任何固体或沉积物。完成蒸馏之后,精馏塔下游的冷阱含有5g物质,经定性分析,该物质含有Si和N。
基于所用一氯硅烷,蒸馏分离的TSA的收率为68%,所得TSA的纯度高于99.5重量%。
将仍然位于搅拌式高压釜中的甲苯、氯化铵、少量TSA、DSA和聚硅氮烷的溶液排出并过滤。经过滤的甲苯含有9g TSA、0.8g DSA、3g聚硅氮烷,不含氯化铵。经干燥的氯化铵滤饼含有3g硅。
实施例1
将3400ml甲苯和466g一氯硅烷先后装入已经提前用惰性气体吹扫过的5l搅拌式高压釜中,该高压釜具有冷却和加热模式并且与蒸馏单元连接,蒸馏单元包含蒸馏塔和冷凝器。以7小时5分钟的时长向该反应溶液中添加177g氨。添加期间,温度恒定在+10℃。添加期间的压力从2.8巴绝对压力升至3.1巴绝对压力。
添加氨之后,将混合物在+10℃下进一步搅拌1小时。然后将反应器溶液调节至-20℃并保持,继续进一步搅拌整夜。
第二天,通过蒸馏单元下游连接的真空泵将压力设定为0.5巴绝对压力并将搅拌式高压釜加热。利用蒸馏单元,将TSA、DSA、甲苯馏分和痕量氯化铵蒸馏分出;合成过程的过量氨经真空泵流入蒸馏的废气。蒸馏液冷凝器的低温恒温器在-20℃的流动温度下操作。完成上述氨向反应溶液的添加之后19小时25分钟,收集到第一滴TSA蒸馏物。收集到第一滴TSA蒸馏物之后3小时50分钟停止蒸馏。蒸馏期间蒸馏液冷凝器的内部过程温度从-3℃升至+3℃。
在蒸馏液冷凝器内-3℃升至+3℃的内部过程温度下,TSA和少量生成的DSA并未定量地凝出。
为了彻底收集未凝出的TSA和DSA,在真空泵的下游安装两个洗涤瓶,瓶中总共装有3370g 20重量%浓度的氢氧化钠溶液。进入洗涤瓶的TSA和DSA被水解。定量分析洗涤瓶内容物,得到15.2g硅。由于水解,不能测定TSA与DSA之间的质量比。如果15.2g的硅全部源自TSA,那么可以推算出收集在洗涤瓶中的TSA量为19.4g。
将收集到的蒸馏溶液过滤,然后除去氯化铵即变得澄清。采用气相色谱定量分析,该溶液相应地含有DSA(12.8g)、TSA(165.7g)和甲苯(145.3g)。进一步用1HNMR定量分析,该溶液相应地含有DSA(14.9g)、TSA(165.5g)和甲苯(143.4g)。
基于所用一氯硅烷,存在于蒸馏溶液中的TSA的收率为66%。将仍然位于搅拌式高压釜中的甲苯、氯化铵、少量TSA、DSA和聚硅氮烷的溶液排出并过滤。经过滤的甲苯含有4g TSA、0.3g DSA、4g聚硅氮烷,不含氯化铵。经干燥的氯化铵滤饼含有3g硅。
参考文献
[1]A.Stock,K.Somieski,Berichte der Deutschen Chemischen Gesellschaft 54 1921pp.740-758
[2]U.Wannagat,Fortschritte der Chemischen Forschung 9(1)1967pp.102-144
[3]A.MacDiarmid,Advances in inorganic chemistry and radiochemistry 3 1961pp.207-256,
[4]B.J.Aylett,M.J.Hakim,Inorganic Chemistry 5(1)1966p.167
[5]R.L.Wells,R.Schaeffer,Journal of the American Chemical Society 88(1)1966pp.37-42
[6]G.D.Miller,WO 2010/141551 A1
[7]C.J.Ritter,III,US 2013/0216463 A1
[8]A.V.Korolev,US 2013/0209343 A1
[9]C.-F.Hoppe,H.Rauleder,Ch.DE102011088814.4
[10]C.-F.Hoppe,H.Rauleder,Ch.G.Uehlenbruck,DE 102012214290.8
[11]C.-F.Hoppe,Ch.G.Uehlenbruck,H.Rauleder,DE 102013209802.2
[12]N.Y.Kovarsky,D.Lubomirsky,US 2011/0136347 A1
- 还没有人留言评论。精彩留言会获得点赞!