非均相双组分的加氢催化剂和氢化丁腈橡胶及其制备方法与流程
本发明涉及一种非均相双组分的加氢催化剂和氢化丁腈橡胶及其制备方法,属于催化加氢制备特种橡胶的领域。
背景技术:
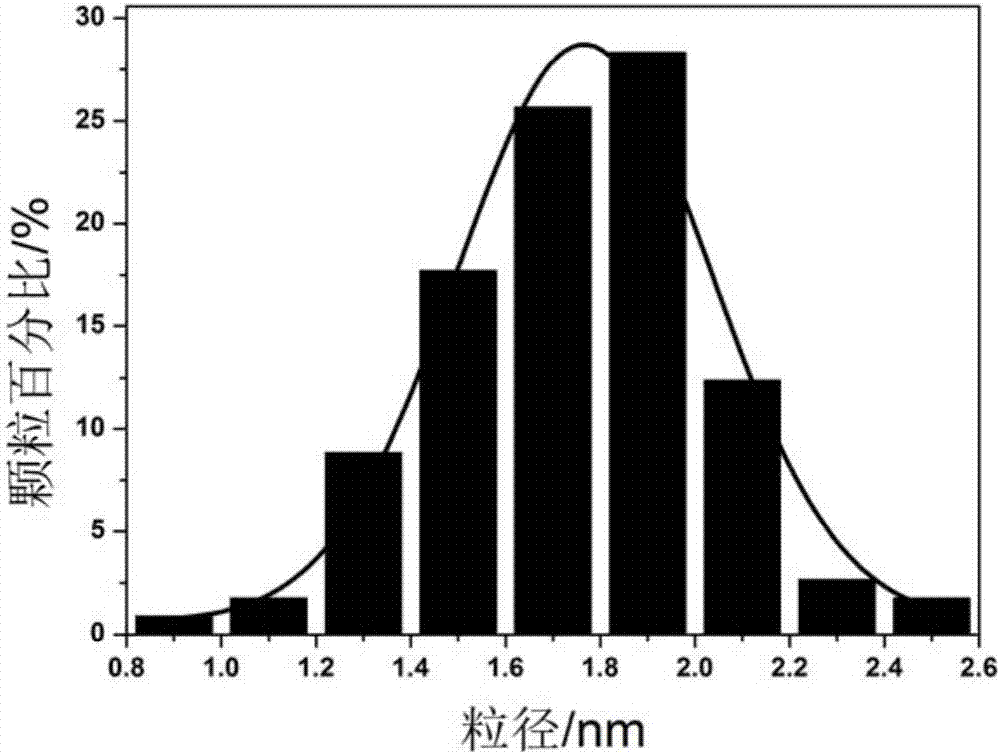
:丁腈橡胶(nbr)称为丁二烯-丙烯腈橡胶,由丁二烯和丙烯腈通过乳液共聚法制得,是目前广泛应用的主要合成橡胶品种之一。nbr分子链中存在腈基,使其具有可耐非极性油品和低芳烃类溶剂的特性,但大量不饱和c=c的存在使其耐热性、耐氧化性能、抗老化性和耐臭氧性能较差。在保留腈基的前提下,对不饱和c=c进行选择性加氢饱和,可以制备高性能和高附加值的氢化丁腈橡胶(hnbr)。hnbr继承了nbr的优点,并且具有优良的耐化学腐蚀性、优异的耐臭氧性、较高的抗压缩永久变形性能等特点,综合性能极为优异,在汽车、航空航天、石油化工等行业领域得到广泛应用。目前,由nbr制取hnbr的加氢工艺主要有溶液加氢法、乳液加氢法和乙烯-丙烯腈共聚法。其中,溶液加氢法的应用最为广泛。根据所用催化剂不同,溶液加氢法又可分为均相溶液加氢和非均相溶液加氢。在均相溶液加氢中,反应物nbr与催化剂在同一相中进行加氢反应,两者可以充分接触,无扩散传质影响,因此均相加氢催化剂所需反应条件较为温和。均相加氢所用催化剂一般为过渡金属类,包括铑(rh)系催化剂、钌(ru)系催化剂、钯(pd)系催化剂和rh-ru双金属催化剂。其中,以rhcl(pph3)3为代表的rh系催化剂最为高效,应用最为广泛。然而,由于均相催化剂与加氢产品分离难度较大,催化剂分离设备的投入大幅提高生产成本。同时,残留在加氢产品中的催化剂造成贵金属资源浪费,并且会加速橡胶老化,缩短产品使用周期。而非均相加氢体系可以有效地避免这类问题,只需简单的过滤或离心操作即可实现催化剂与产物的分离,使贵金属催化剂可重复利用,因此从长远来看,发展非均相催化加氢体系更具有工业前景和实用价值。技术实现要素:为解决上述技术问题,本发明的目的是提供一种非均相双组分的加氢催化剂及其制备方法,该方法选择合适的助剂组分,能够提高贵金属活性组分的催化活性,两组分之间存在“协同效应”,可以减少贵金属用量,降低催化剂成本,从而降低制备氢化丁腈的成本。本发明还提供了一种采用上述非均相双组分的加氢催化剂制备的氢化丁腈橡胶及其制备方法。为了达到上述目的,本发明提供了一种非均相双组分的加氢催化剂的制备方法,该制备方法具体包括以下步骤:利用偶联剂对二氧化硅空心微球(shms)载体进行氨基修饰,然后,在所述载体上负载活性组分m1和助剂组分m2,经干燥、h2还原处理,得到非均相双组分的加氢催化剂;其中,所述活性组分m1选自pd或rh,优选为pd,所述助剂组分m2选自zr或ti,优选为zr。在上述加氢催化剂的制备方法中,优选采用化学键合法负载所述活性组分m1,采用溶剂挥发法负载所述助剂组分m2。二氧化硅空心微球载体与偶联剂进行水解、缩水反应,得到表面修饰氨基的载体,然后,以活性组分m1的盐溶液为活性组分m1的前体液,将修饰后的载体与活性组分m1的盐溶液反应,利用载体表面的氨基与活性金属m1之间的络合作用,负载一定量的活性金属m1,然后,以助剂组分m2的盐溶液为助剂组分m2的前体液,将负载m1的载体加入助剂组分m2的盐溶液中,采用溶剂挥发法负载助剂组分m2,最后,经干燥、h2还原等处理,得到非均相双组分的加氢催化剂m1-m2/shms。在加氢催化剂的制备过程中,化学键合法和溶剂挥发法联合用于负载双组分,可以使活性金属和组分金属的分散和牢固负载,保证反应后不流失。在上述加氢催化剂的制备方法中,优选地,所采用的活性金属m1的盐包括mclx或m(oac)x溶液;所采用的助剂组分m2的盐包括mclx或m(oc3h7)4或m(oc4h9)4溶液。在上述加氢催化剂的制备方法中,优选地,所述二氧化硅空心微球的粒径为2-20μm,比表面积为50-500m2/g。在上述加氢催化剂的制备方法中,优选地,所述偶联剂选自氨基硅烷偶联剂或巯基硅烷偶联剂,更优选为氨基硅烷偶联剂,例如n-(β-氨乙基)-γ-氨丙基三甲氧基硅烷。在上述加氢催化剂的制备方法中,优选地,所述活性组分m1与助剂组分m2的原子比为10-0.5:1,更优选为5-1:1。在上述加氢催化剂的制备方法中,优选地,以所述加氢催化剂的总量为100wt%计,活性组分m1和助剂组分m2的总负载量为1-8wt%。本发明还提供了一种采用上述方法制备的非均相双组分的加氢催化剂。本发明还提供一种由上述非均相双组分的加氢催化剂制备氢化丁腈橡胶的制备方法,该制备方法包括以下步骤:用100ml的有机溶剂溶解1-8g丁腈橡胶,得到胶液;将上述的加氢催化剂以0.1-5g/100ml有机溶剂的添加量加入到所述胶液中,进行催化加氢反应;对催化加氢后的胶液进行离心分离,回收加氢催化剂,蒸发有机溶剂得到氢化丁腈橡胶。在上述氢化丁腈橡胶的制备方法中,优选地,催化加氢反应的温度为50-100℃,氢气压力为1-10mpa,反应时间为1-12h。在上述氢化丁腈橡胶的制备方法中,优选地,丁腈橡胶的分子量为1000-100000;更优选地,所述丁腈橡胶的分子量为1000-50000。在上述氢化丁腈橡胶的制备方法中,有机溶剂可以是丙酮,或其他容易挥发的有机溶剂。在上述氢化丁腈橡胶的制备方法中,nbr的加氢度达到90%以上,选择性可达100%。在上述氢化丁腈橡胶的制备方法中,优选地,所述二氧化硅空心微球是通过以下步骤制备的:内层水相的配制:将7-21g水玻璃、11-33g去离子水混合,得到内层水相,所述水玻璃中sio2的含量为26wt%;油相的配制:将72ml正己烷、1.5gtween80、1.5gspan80混合,得到油相;外层水相的配制:将无机盐与250ml去离子水混合,得到浓度为0.3-3mol/l的外层水相;所采用的无机盐可以为nh4hco3、nh4cl、nh4no3或(nh4)2so4等;将内层水相与油相混合,以6000-14000rpm的转速搅拌乳化1min,形成水/油体系;将水/油体系与外层水相混合,在搅拌条件下于常温反应2h,然后经过过滤、去离子水及乙醇洗涤、充分干燥后,制备得到所述二氧化硅空心微球。本发明还提供了一种采用上述方法制备的氢化丁腈橡胶。通过本发明的非均相双组分的加氢催化剂的制备方法得到的催化剂中,活性组分分散性好,颗粒尺寸可控,贵金属颗粒能稳定牢固地负载于载体上,反应后不会发生团聚和流失,并且活性组分与助剂组分间的协同作用可以促进催化剂的加氢性能,可降低活性金属使用量,能够在提高催化剂的可利用度并保证hnbr的产品性能的同时降低催化剂的合成成本。本发明的方法所制备的非均相双组分加氢催化剂的催化活性高、选择性高、稳定性好,制备成本相对较低且加氢条件温和、工艺简单。本发明所提供的氢化丁腈橡胶的制备方法是利用上述非均相双组分的加氢催化剂完成的,该制备方法的加氢度高,而且选择性可以达到100%。附图说明图1为实施例3、6、8所采用的m1-m2/shms催化剂的tem图;图2为实施例3、6、8所采用的m1-m2/shms催化剂的活性组分颗粒粒径分布图;图3为实施例3制备的hnbr与nbr红外光谱图;图4为实施例3制备的hnbr与nbr的1hnmr谱图;图5为实施例3、6、8所采用的m1-m2/shms催化剂用于nbr加氢反应后的tem图;图6为实施例3、6、8所采用的m1-m2/shms催化剂用于nbr加氢反应后的活性组分颗粒粒径分布图;图7为单组分pd催化剂(a)和双组分pd-zr(原子比3:1)催化剂(b)的xps图。具体实施方式为了对本发明的技术特征、目的和有益效果有更加清楚的理解,现对本发明的技术方案进行以下详细说明,但不能理解为对本发明的可实施范围的限定。在以下实施例中,合成所用原料中水玻璃为工业级,其他均为试剂级;tem图像由日本jem2100lab6透射电子显微镜获得;红外谱图由美国magna-ir560e.s.p型ft-ir光谱仪获得;1hnmr谱图由日本jnm-la300ft-nmr核磁共振波谱仪获得;加氢后得到的hnbr中pd含量的测定由optima7300v型电感耦合等离子光谱仪获得。加氢度通过红外表征结果计算得到。ft-ir表征可检测nbr及加氢产物hnbr中c=c的含量,并通过公式进行氢化度的计算。如表1所示,表1列出了图3中的红外光谱图的主要特征峰,hnbr氢化度可通过如下过程计算:式中a(723)为723cm-1处的吸收峰强度,a(970)和a(2236)同理。式中k为hnbr的特征常数,k(723)=0.25,k(970)=2.3。hnbr氢化度(mol%)=100-c(nbr)/[c(nbr)+c(hnbr)]×100表1nbr及hnbr红外特征峰波数(cm-1)基团种类峰强度变化3500r-nh2或r2-nh-c≡n若被加氢饱和后新出现的峰2236-c≡n氢化度计算过程中以此为内标9701,4-反式,-ch=ch-加氢后峰强度减弱或消失9171,2-双键,-ch=ch2加氢后峰强度减弱或消失7501,4-顺式,-ch=ch-加氢后峰强度减弱或消失723-[ch2]n-中的c-h键(n4)c=c被加氢饱和后新出现的峰本发明的具体实施例提供了非均相双组分的加氢催化剂的制备方法,该制备方法具体包括以下步骤:对于shms的修饰可以按照以下步骤进行,但不限于此:取1gshms在100℃下烘干,冷却到室温,分散在一定溶剂中;将一定量n-(β-氨乙基)-γ-氨丙基三甲氧基硅烷溶解,制备前体液;将前体液和shms分散液混合,在室温下搅拌10min后,转移至80℃恒温水浴锅反应24h;待冷却至室温,离心分离,用溶剂洗涤以除去载体表面未反应的物质;洗涤后将样品分散在乙醇中,在60℃水浴锅中搅拌8h,冷却至室温,分离,在100℃烘箱内烘干,得到氨基修饰的shms。双组分催化剂的合成可以按照以下步骤进行,但不限于此:取氨基修饰的shms烘干,冷却后分散在适量甲苯中;采用化学键合法负载m1:将适量活性组分m1的盐溶解在适量溶剂(例如ch2cl2)中,作为活性组分前体液;将前体液逐滴加入分散有载体的甲苯溶液中;混合液在适当温度(例如60℃)恒温水浴锅中反应适当时间(例如12h),得到负载活性组分m1的催化剂;然后将其分散于适量乙醇中,磁力搅拌适当时间至其分散均匀;采用溶剂挥发法负载助剂组分m2:取适量助剂组分m2的盐(此量根据活性组分m1与助剂组分m2的原子比推算)溶于适量乙醇中,得到助剂组分前体液;将助剂组分前体液逐滴滴入混合液中,继续搅拌适当时间;在适当温度(例如60℃)水浴加热适当时间(例如24h)后,离心分离,干燥,得到双组分催化剂。本发明的具体实施例还提供了氢化丁腈橡胶的制备方法,但不限于此。实施例1本实施例提供了一种非均相双组分的加氢催化剂的制备方法,其包括以下步骤:采用n-(β-氨乙基)-γ-氨丙基三甲氧基硅烷氨基修饰二氧化硅空心微球shms,然后,以氨基修饰的shms作为载体,以pd(oac)2溶液为活性组分的前体液,采用化学键合法负载pd,以zr(c3h7o)4溶液为助剂组分的前体液,采用溶剂挥发法负载zr,制备总负载量为7wt%,pd:zr原子比为7:1的非均相双组分的加氢催化剂pd-zr/shms。本实施例还提供了hnbr的制备方法,该制备方法包括以下步骤:将1gnbr溶解在100ml丙酮溶剂中得到胶液,取1g非均相双组分的加氢催化剂pd-zr/shms加入到胶液中,在高压加氢反应釜中进行催化加氢反应,反应条件为:温度60℃,氢气压力4mpa,反应时间6h;反应结束后,对非均相催化剂进行离心分离回收,对胶液进行蒸发以去除有机溶剂,得到产品hnbr。该方法的加氢度、选择性及存在于制备的hnbr中的pd含量列于表2中。实施例2本实施例提供了一种非均相双组分的加氢催化剂的制备方法,其包括以下步骤:采用n-(β-氨乙基)-γ-氨丙基三甲氧基硅烷氨基修饰二氧化硅空心微球shms,然后,以氨基修饰的shms作为载体,以pd(oac)2溶液为活性组分的前体液,采用化学键合法负载pd,以zr(c3h7o)4溶液为助剂组分的前体液,采用溶剂挥发法负载zr,最后,经干燥、h2还原等处理,制备得到总负载量为7wt%,pd:zr原子比为5:1的非均相双组分的加氢催化剂pd-zr/shms。hnbr的制备方法如实施例1,该方法的加氢度、选择性及存在于制备的hnbr中的pd含量列于表2中。实施例3本实施例提供了一种非均相双组分的加氢催化剂的制备方法,其包括以下步骤:采用n-(β-氨乙基)-γ-氨丙基三甲氧基硅烷氨基修饰二氧化硅空心微球shms,然后,以氨基修饰的shms作为载体,以pd(oac)2溶液为活性组分的前体液,采用化学键合法负载pd,以zr(c3h7o)4溶液为助剂组分的前体液,采用溶剂挥发法负载zr,最后,经干燥、h2还原等处理,制备得到总负载量为7wt%,pd:zr原子比为3:1的非均相双组分的加氢催化剂pd-zr/shms。hnbr的制备方法如实施例1,该方法的加氢度、选择性及存在于制备的hnbr中的pd含量列于表2中。实施例4本实施例提供了一种非均相双组分的加氢催化剂的制备方法,其包括以下步骤:采用n-(β-氨乙基)-γ-氨丙基三甲氧基硅烷氨基修饰二氧化硅空心微球shms,然后,以氨基修饰的shms作为载体,以pd(oac)2溶液为活性组分的前体液,采用化学键合法负载pd,以zr(c3h7o)4溶液为助剂组分的前体液,采用溶剂挥发法负载zr,最后,经干燥、h2还原等处理,制备得到总负载量为7wt%,pd:zr原子比为1:1的非均相双组分的加氢催化剂pd-zr/shms。hnbr的制备方法如实施例1。该方法的加氢度、选择性及存在于制备的hnbr中的pd含量列于表2中。实施例5本实施例提供了一种非均相双组分的加氢催化剂的制备方法,其包括以下步骤:采用n-(β-氨乙基)-γ-氨丙基三甲氧基硅烷氨基修饰二氧化硅空心微球shms,然后,以氨基修饰的shms作为载体,以pd(oac)2溶液为活性组分的前体液,采用化学键合法负载pd,以zr(c3h7o)4溶液为助剂组分的前体液,采用溶剂挥发法负载zr,最后,经干燥、h2还原等处理,制备得到总负载量为5wt%,pd:zr原子比为3:1的非均相双组分的加氢催化剂pd-zr/shms。hnbr的制备方法如实施例1,该方法的加氢度、选择性及存在于制备的hnbr中的pd含量列于表2中。实施例6本实施例提供了一种非均相双组分的加氢催化剂的制备方法,其包括以下步骤:采用n-(β-氨乙基)-γ-氨丙基三甲氧基硅烷氨基修饰二氧化硅空心微球shms,然后,以氨基修饰的shms作为载体,以pd(oac)2溶液为活性组分的前体液,采用化学键合法负载pd,以zr(c3h7o)4溶液为助剂组分的前体液,采用溶剂挥发法负载zr,最后,经干燥、h2还原等处理,制备得到总负载量为7wt%,pd:zr原子比为3:1的非均相双组分的加氢催化剂pd-zr/shms。本实施例还提供了hnbr的制备方法,该方法包括以下步骤:将1gnbr溶解在100ml丙酮溶剂中得到胶液,取0.5g非均相双组分的加氢催化剂pd-zr/shms加入到胶液中,在高压加氢反应釜中进行催化加氢反应。反应条件为:温度60℃,氢气压力4mpa,反应时间6h;反应结束后,对非均相催化剂进行离心分离回收,对胶液进行蒸发以除去有机溶剂,得到产品hnbr。该方法的加氢度、选择性及存在于制备的hnbr中的pd含量列于表2中。实施例7本实施例提供了一种非均相双组分的加氢催化剂的制备方法,其包括以下步骤:采用n-(β-氨乙基)-γ-氨丙基三甲氧基硅烷氨基修饰二氧化硅空心微球shms,然后,以氨基修饰的shms作为载体,以pd(oac)2溶液为活性组分的前体液,采用化学键合法负载pd,以zr(c3h7o)4溶液为助剂组分的前体液,采用溶剂挥发法负载zr,最后,经干燥、h2还原等处理,制备得到总负载量为5wt%,pd:zr原子比为3:1的非均相双组分的加氢催化剂pd-zr/shms。本实施例还提供了hnbr的制备方法,该方法包括以下步骤:将1gnbr溶解在100ml丙酮溶剂中得到胶液,取0.5g非均相双组分的加氢催化剂pd-zr/shms加入到胶液中,在高压加氢反应釜中进行催化加氢反应。反应条件为:温度60℃,氢气压力4mpa,反应时间6h,反应结束后,对非均相催化剂进行离心分离回收,对胶液进行蒸发以除去有机溶剂,得到产品hnbr。该方法的加氢度、选择性及存在于制备的hnbr中的pd含量列于表2中。实施例8本实施例提供了一种非均相双组分的加氢催化剂的制备方法,其包括以下步骤:采用n-(β-氨乙基)-γ-氨丙基三甲氧基硅烷氨基修饰二氧化硅空心微球shms,然后,以氨基修饰的shms作为载体,以pd(oac)2溶液为活性组分的前体液,采用化学键合法负载pd,以zr(c3h7o)4溶液为助剂组分的前体液,采用溶剂挥发法负载zr,最后,经干燥、h2还原等处理,制备得到总负载量为7wt%,pd:zr原子比为3:1的非均相双组分的加氢催化剂pd-zr/shms。本实施例还提供了hnbr的制备方法,该方法包括以下步骤:将1gnbr溶解在100ml丙酮溶剂中得到胶液,取0.1g非均相双组分的加氢催化剂pd-zr/shms加入到胶液中,在高压加氢反应釜中进行催化加氢反应。反应条件为:温度60℃,氢气压力4mpa,反应时间6h,反应结束后,对非均相催化剂进行离心分离回收,对胶液进行蒸发以除去有机溶剂,得到产品hnbr。该方法的加氢度、选择性及存在于制备的hnbr中的pd含量列于表2中。实施例9本实施例提供了一种非均相双组分的加氢催化剂的制备方法,其包括以下步骤:采用n-(β-氨乙基)-γ-氨丙基三甲氧基硅烷氨基修饰二氧化硅空心微球shms,然后,以氨基修饰的shms作为载体,以pd(oac)2溶液为活性组分的前体液,采用化学键合法负载pd,以ti(c3h7o)4溶液为助剂组分的前体液,采用溶剂挥发法负载ti,最后,经干燥、h2还原等处理,制备得到总负载量为7wt%,pd:ti原子比为5:1的非均相双组分的加氢催化剂pd-ti/shms。本实施例还提供了hnbr的制备方法,该方法包括以下步骤:将1gnbr溶解在100ml丙酮溶剂中得到胶液,取1g非均相双组分的加氢催化剂pd-ti/shms加入到胶液中,在高压加氢反应釜中进行催化加氢反应。反应条件为:温度60℃,氢气压力4mpa,反应时间6h,反应结束后,对非均相催化剂进行离心分离回收,对胶液进行蒸发以除去有机溶剂,得到产品hnbr。该方法的加氢度、选择性及存在于制备的hnbr中的pd含量列于表2中。实施例10本实施例提供了一种非均相双组分的加氢催化剂的制备方法,其包括以下步骤:采用n-(β-氨乙基)-γ-氨丙基三甲氧基硅烷氨基修饰二氧化硅空心微球shms,然后,以氨基修饰的shms作为载体,以pd(oac)2溶液为活性组分的前体液,采用化学键合法负载pd,以ti(c3h7o)4溶液为助剂组分的前体液,采用溶剂挥发法负载ti,最后,经干燥、h2还原等处理,制备得到总负载量为7wt%,pd:ti原子比为3:1的非均相双组分的加氢催化剂pd-ti/shms。本实施例还提供了hnbr的制备方法,如实施例9,该方法的加氢度、选择性及存在于制备的hnbr中的pd含量列于表2中。试验例本试验例对实施例3、6、8所制备的pd:zr原子比为3的pd-zr/shms催化剂进行了tem表征(如图1所示),由图1可知,上述实施例制得的加氢催化剂的活性组分颗粒分布均匀,而且颗粒尺寸小,由粒径统计图(图2)可知,颗粒尺寸大约集中在1.8nm。图5是实施例3、6、8所制备的pd:zr原子比为3的pd-zr/shms催化剂用于nbr加氢反应后的tem表征,说明利用本发明提供的负载方法得到的催化剂在反应后活性组分颗粒仍然分布均匀,颗粒尺寸小,由粒径统计图(图6)可知,颗粒尺寸大约集中在2.0nm。本试验例中,还对实施例3所制备的hnbr与nbr进行了红外谱图检测,结果如图3所示。由图3可知,-cn在2230.0cm-1吸收峰强度始终未减弱,在3500.0cm-1的-nh2吸收峰并未出现,说明反应前后-cn没有发生变化,即-cn没有被加氢,选择性为100%;在971.3cm-1及920.0cm-1处的1,4-双键基团和1,2-双键基团的吸收峰不同程度地减弱,甚至消失,说明双键基本被完全加氢。同时,加氢后在723.1位置出现新的峰,属于-[ch2]n-(其中n>4)结构,即为分子链中c=c加氢饱和后新出现的结构。图4为实施例3所制备的hnbr与nbr的1hnmr谱图。由图4可知,通过红外数据计算得到的加氢度为96.2%的hnbr,在化学位移为5.53、5.37的-ch=ch-,5.09、4.97的-c-c=ch2,2.27、2.09的-c-ch2-c=c-以及1.64的-c-ch2-c-c-c=c-质子吸收峰完全消失。新增了化学位移分别在1.57、1.29、0.86,代表饱和烷烃链段的质子吸收峰。通过核磁数据计算得到的加氢度为97.5%,可以确定红外数据计算加氢度的正确性。对比例1本对比例提供一种仅用pd作为活性组分的非均相加氢催化剂,催化nbr加氢制备hnbr,以便与实施例1-4进行对比。采用n-(β-氨乙基)-γ-氨丙基三甲氧基硅烷氨基修饰二氧化硅空心微球shms作为载体,以pd(oac)2溶液为活性组分的前体液,负载量为7%,得到单金属催化剂pd/shms。按照实施例1中的nbr加氢实验条件进行反应,相同的反应条件下nbr加氢度为92.1%。对比例2本对比例提供一种采用等体积共浸渍法制备的pdzr非均相双组分的加氢催化剂,催化nbr加氢制备hnbr,以便与实施例3进行对比。以二氧化硅空心微球为载体,以pdcl2溶液为活性组分的前体液,以zr(c3h7o)4溶液为助剂组分的前体液,最后,经干燥、h2还原等处理,制备得到总负载量为7%,pd-zr原子比为3的非均相双组分的加氢催化剂pd-zr/shms。按照实施例1中的nbr加氢实验条件进行反应,相同的反应条件下nbr加氢度为88.0%。但icp检测存在于所制备的hnbr产品中的pd含量为355.0ppm,表明此法制备的催化剂中pd流失严重。对比例3本对比例提供一种采用等体积浸渍法制备的zr基催化剂,催化nbr加氢制备hnbr,以便与实施例1-4进行对比。以二氧化硅空心微球为载体,以zr(c3h7o)4溶液为前体液,制备总负载量为7%的zr催化剂。按照实施例1中的nbr加氢实验条件进行反应,相同的反应条件下nbr几乎没有加氢。表2实施例1-10和对比例1-3的加氢度、选择性及存在于制备的hnbr中的pd含量通过以上实施例以及表2可以看出,利用本发明提供的催化剂对丁腈橡胶进行选择性非均相加氢的加氢度高,而且选择性可以达到100%,残存在hnbr产品中的pd含量极少。单组分pd催化剂和双组分pd-zr(原子比3:1)催化剂的xps图以及相关数据分别如图7和表3所示。表3根据图7和表3的内容可以看出,pd03d5/2和pd03d3/2对应的能谱峰位置均向低结合能方向移动了0.2ev,这是因为助剂组分zr的引入对pd周围电子环境产生了影响,使得pd周围的电子状态发生偏移,证明双组分之间存在协同效应。综上所述,采用本发明的非均相双组分的加氢催化剂制备氢化丁腈橡胶,能够提高贵金属活性组分的催化活性,两组分之间存在“协同效应”,可以减少贵金属用量,降低催化剂成本,从而降低制备氢化丁腈的成本。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1