加氢异构化催化剂及其制备方法和加氢裂化尾油的加氢处理方法与流程
本发明涉及加氢裂化尾油的加氢处理领域,具体涉及加氢异构化催化剂及其制备方法和加氢裂化尾油的加氢处理方法。
背景技术:
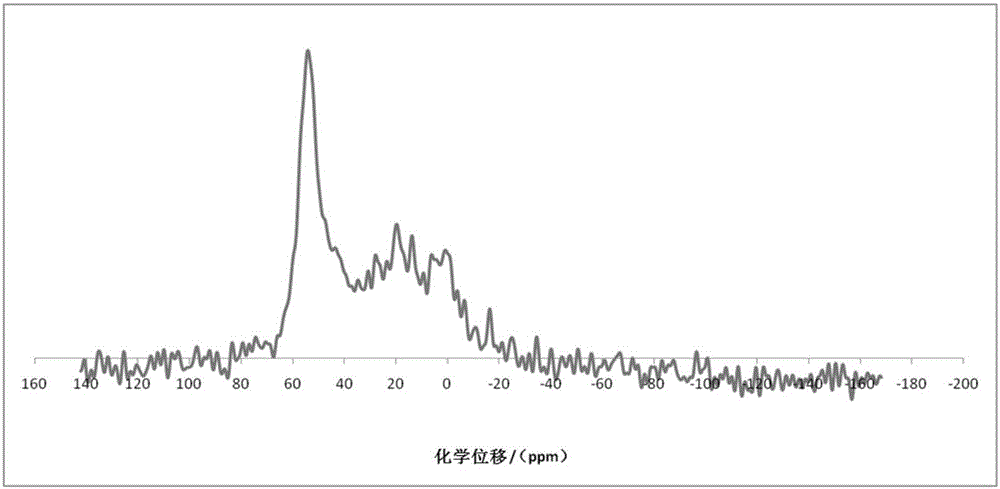
:富含长链烷烃的原料主要包括加氢裂化尾油、植物油、费-托合成蜡和废润滑油等,它们的凝点较高,低温流动性差,在储存、运输和使用的过程中,随着环境温度的降低会使油品的流动性变差,甚至凝固,从而堵塞容器和管道和中断发动机供油。目前解决含蜡质油因凝点过低引起的各种问题的方法有很多,主要包括溶剂脱蜡、催化脱蜡和异构脱蜡。溶剂脱蜡就是利用蜡在溶剂中的溶剂性能来除去,这种方法的缺点是溶剂选择困难、浪费大量的有机溶剂、对人体有害并且污染环境、设备投资和操作费用高,以及产品质量受原料限制。催化脱蜡就是利用具有择形裂解功能的催化剂,使馏分中的蜡组分发生选择性裂化,生成小分子烃类,例如美国专利申请us4247388和us4659311就是使用催化脱蜡的方法除去润滑油中的蜡,这种方法的缺点是由于把大量的大分子化合物转化成小分子物质,使基础油收率低、粘度指数损失大、副产物的附加值低。与前两种脱蜡方法相比,异构脱蜡是使大分子蜡发生加氢异构化反应生成具有更低凝点的异构烷烃,从而达到降低凝点、改善基础油粘温性质和低温流动性性质的作用,而所生成的异构烷烃仍然存在于基础油中,可以得到较高的基础油收率。目前关于异构脱蜡催化剂的报道很多,例如,美国专利申请us4518485、us5990371、us5135638、us4419420、us5110445等都报道了涉及异构脱蜡技术生产润滑油基础油,其中使用的酸性组分的主要有mor、zsm-22、zsm-23、zsm-48、sapo-11、sapo-31、sapo-41、nu-10和kz-2等,这些材料都能够在一定程度上使石蜡烃发生异构化反应,但由于自身分子筛的特点,只能使反应物中的一部分发生异构化反应,而其余发生裂解反应,而异构程度越大,发生裂解反应就越多,使产物的收率下降。美国专利申请us5282958公开了一种用于异构脱蜡的催化剂。该催化剂中含有中孔分子筛,如zsm-5、zsm-22、zsm-23、zsm-11等。该专利申请中所用分子筛晶体尺寸小于0.5μm,孔尺寸为之间。由于对所用分子筛晶体尺寸以及孔尺寸进行了限定,在专利规定工艺条件下异构脱蜡催化剂具有较好的降凝能力。美国专利申请us7482300、us5075269公开了一种含有zsm-48的异构催化剂。该专利申请对zsm-48的合成条件进行了限定。通过限定的合成条件,zsm-48分子筛具有较高的硅铝氧化物分子比且形貌可控。美国专利申请us8513150公开了一种含有介孔的y型分子筛。在该专利申请中y型分子筛先在低温下焙烧随后在含有水蒸汽的气体里高温(1250°f~1450°f)焙烧。经焙烧后分子筛内形成介孔结构,且大介孔对小介孔的比例为5以上。美国专利申请us5397454公开了使用具有小晶粒尺寸和焙烧后氢型的约束指数为13或更大的例如ssz-32的沸石进行加氢转化的方法。催化剂的氧化硅与氧化铝之比大于20且小于40。美国专利申请us5300210也涉及使用ssz-32进行烃转化的方法。美国专利申请us5300210的ssz-32不限定于小晶粒尺寸。美国专利申请us7141529公开了用不同的金属(选自ca、cr、mg、la、ba、pr、sr、k和nd的金属以及第viii族金属)对分子筛进行金属改性的方法,以提供使用nc-16进料时具有改进的异构化选择性的催化剂。该专利申请所用方法为载体成型之后用含有金属离子的液体进行浸渍从而负载改性金属。中国专利申请cn103964458a公开了一种高硅铝比多级孔道的beta沸石及其制备方法。该beta沸石的孔道在2nm以下、5-11nm和50nm以上均有孔径分布,其中,微孔体积为0.19cm3/g以上,介孔和大孔总体积为0.35cm3/g以上,其硅铝比为90以上,比表面积为400m2/g以上。该制备方法包括以下步骤:将原料beta沸石进行第一次酸处理;将第一次酸处理后的beta沸石进行第一次焙烧;将第一次焙烧后的beta沸石进行第二次酸处理,得到所述的高硅铝比多级孔道的beta沸石。该发明的制备方法操作简单高效,制备得到的高硅铝比多级孔道的beta沸石具有很强的酸、热和水热稳定性以及良好的扩散性能。技术实现要素:本发明的目的是提供一种加氢异构化催化剂及其制备方法和加氢裂化尾油的加氢处理方法,采用本发明所述的加氢异构化催化剂进行加氢裂化尾油的加氢处理,所获得的目标产物的黏度指数较高、倾点较低且收率较高。本发明提供了一种加氢异构化催化剂,该催化剂含有载体和负载在所述载体上的活性金属组分,所述活性金属组分为viii族贵金属中的至少两种,所述载体含有十元环硅铝分子筛,所述十元环硅铝分子筛的氧化硅/氧化铝摩尔比为120~300;含有介孔结构且在低温氮气吸附-脱附曲线p/p0=0.4-0.99处出现一个闭合滞后环,且所述闭合滞后环的起始位置在p/p0=0.4-0.7处。本发明还提供了上述加氢异构化催化剂的制备方法,该方法包括以下步骤:(i)采用浸渍法将活性金属组分前驱体和有机络合剂负载在载体上,然后进行干燥、焙烧,得到半成品催化剂,优选地,所述焙烧的条件使得以半成品催化剂的总重量为基准,所述半成品催化剂中炭含量为0.05-0.5重量%,优选为0.1-0.4重量%;(ii)以含有有机络合剂的溶液作为浸渍液,对步骤(i)所得半成品催化剂进行浸渍,然后进行干燥。本发明还提供了一种加氢裂化尾油的加氢处理方法,该方法包括:在加氢异构化反应条件下,将加氢裂化尾油与催化剂接触反应,其中,所述催化剂为本发明所述的加氢异构化催化剂或者根据上述方法制备的加氢异构化催化剂。本发明的发明人经过大量的研究发现,在分子筛的制备过程中,通过对晶化后的母液进行恰当的化学处理,可以制备具有特殊物化性质的十元环硅铝分子筛,具体地,本发明所述的十元环硅铝分子筛具有较高的硅铝比,含有介孔结构,并且分子筛前体中富含五配位铝,而分子筛成品中含有很少的五配位铝,甚至基本上不含五配位铝。在本发明所述的加氢异构化催化剂中,由于其中包含的十元环硅铝分子筛具有特殊的物化性质,使得该加氢异构化催化剂在应用于加氢裂化尾油的加氢处理工艺中时,所获得的目标产物的黏度指数较高、倾点较低且收率较高。附图说明图1是制备例1中制备的分子筛前体c-1的27alnmr图谱。图2是制备例1中制备的分子筛成品h-1的27alnmr图谱。图3是制备例1中制备的分子筛成品h-1的xrd图谱。图4是制备例1中制备的分子筛成品h-1的氮气吸附-脱附曲线图。图5是制备对比例3中制备的分子筛前体dc-3的27alnmr图谱。图6是制备对比例3中制备的分子筛成品dh-3的氮气吸附-脱附曲线图。具体实施方式在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。本发明所述的加氢异构化催化剂含有载体和负载在所述载体上的活性金属组分,所述载体含有十元环硅铝分子筛。本发明所述的十元环硅铝分子筛具有高硅的特点。按照本领域常规方法制备的十元环硅铝分子筛的氧化硅/氧化铝摩尔比一般小于100。本发明所述的十元环硅铝分子筛的氧化硅/氧化铝摩尔比为120~300,具体地,例如可以为120、130、140、150、160、170、180、190、200、210、220、230、240、250、260、270、280、290、300以及这些点值中的任意两个所构成的范围中的任意值。优选情况下,所述十元环硅铝分子筛的氧化硅/氧化铝摩尔比为150~200。本发明所述的十元环硅铝分子筛含有介孔结构。按照本领域常规方法制备的十元环硅铝分子筛通常为微孔分子筛,不含有介孔结构。在本发明所述的十元环硅铝分子筛的低温氮气吸附-脱附曲线上,吸附支和脱附支在p/p0=0.4-0.99处出现一个闭合滞后环,且闭合滞后环的起始位置在p/p0=0.4-0.7处,而现有技术制备的十元环硅铝分子筛则不具有该特征,即在该区间内没有滞后环或滞后环的起始位置出现在较高分压处(通常在p/p0>0.7处)。在优选情况下,所述闭合滞后环的起始位置在p/p0=0.4-0.6处。本发明所述的十元环硅铝分子筛经氮气吸附bet(brunner-emmet-teller)方法表征,所述分子筛中的介孔面积可以为50m2/g~250m2/g,所述分子筛的比表面积可以为150m2/g~400m2/g,介孔面积占比表面积的比例可以为20%~70%,优选为25%~65%。本发明所述的十元环硅铝分子筛的前体中富含五配位铝,而分子筛成品中五配位铝的含量很少,甚至基本上不含五配位铝。具体地,所述十元环硅铝分子筛的前体中五配位铝的含量为4-30重量%,优选为10-30重量%;且该分子筛的成品中五配位铝的含量为3重量%以下,优选为2重量%以下,更优选为1重量%以下,最优选为不含五配位铝。通常,含铝分子筛制备可以分为成胶、晶化、后处理等步骤。为了获得具有高硅、含有介孔的十元环硅铝分子筛,需要在含铝分子筛合成过程中的后处理步骤经特殊处理。在优选情况下,所述十元环硅铝分子筛按照以下步骤制备:(1)将晶化后的母液进行过滤,形成干基含量为5-30%的滤饼;(2)将所述滤饼直接进行焙烧,得到分子筛前体;(3)将所述分子筛前体进行水热处理;(4)将水热处理产物进行过滤、洗涤、干燥处理。在步骤(1)中,对晶化后的母液进行过滤的目的是去除合成母液。本发明对过滤形成的滤饼的干基含量进行了特别的限定。具体地,所述滤饼中干基含量为5-30%,优选为6-15%。当所述滤饼中干基含量不在上述范围内时,所制备的分子筛的物化性质不在本发明限定的范围之内。在本发明中,“干基”定义为:在600℃焙烧4小时后物质质量比上焙烧前物质质量的百分比。在步骤(2)中,步骤(1)形成的滤饼直接在高温下进行焙烧而不进行烘干处理。在本发明中,所述焙烧的温度可以为400-600℃,优选为450-550℃。焙烧时升温速率可以为10℃/分钟~100℃/分钟,优选为20℃/分钟~50℃/分钟。所述焙烧的时间可以为1小时~12小时,优选为2小时~6小时。所述焙烧的环境可以为自然环境,即焙烧时不需特意通入含氧气体。由于即使是在自然环境下焙烧,滤饼中的水能够把模板剂氧化掉,同时也可以与分子筛中的铝进行作用从而形成非骨架铝。特别地,本发明中通过步骤(2)处理的产物(即分子筛前体)含有大量的五配位非骨架铝(即五配位铝)。五配位非骨架铝的定义为在27alnmr图谱中化学位移б为15~40ppm的峰。27alnmr图谱测量条件可以参见公开文献,如guoliangzhaoetal,appliedcatalysisa:general299(2006)167–174。在本发明中,步骤(2)处理的产物(即分子筛前体)中五配位铝的含量为4-30重量%,优选为10-30重量%。在步骤(2)中,焙烧处理后样品可以自然冷却降温,目标温度优选为室温。在步骤(3)中,所述水热处理的介质优选为酸性水。在本发明中,所述酸性水是指含h+的h2o溶液。其中,h2o为常规方法获得称为“水”的液体物质。h+为有机酸和/或无机酸解离释放的离子。为了获得所述酸性水,可以在“水”中添加盐酸、硫酸、磷酸、硝酸、柠檬酸、乙酸、马来酸、草酸、氨基三乙酸、1,2-环己烷二胺四乙酸、柠檬酸、酒石酸和苹果酸中的至少一种,优选为盐酸和/或柠檬酸。所述酸性水中无机酸和/或有机酸的含量可以为0.1m~5m,优选为0.2m~2m。在步骤(3)中,所述水热处理的液固体积比可以为5-200,优选为20-100。在步骤(3)中,所述水热处理的温度可以为100℃~300℃,优选为100℃~200℃。在步骤(3)中,所述水热处理的时间可以为0.5小时~24小时,优选为1小时~12小时,更优选为1小时~4小时。在步骤(3)中,所述水热处理优选在密闭容器中实施,且所述水热处理的压力优选为密闭容器在水热条件下形成的自压。在步骤(4)中,分子筛经步骤(3)处理后需要经过滤、洗涤。其中,过滤方法可以为本领域技术人员所熟知的方法。所述洗涤的的过程可以为用去离子水进行水洗,水洗直到滤液ph值为6~8结束,优选为6~7。溶液的ph值测量可以用ph试纸或ph计进行测量,测量方法为本领域技术人员所熟知的方法。在步骤(4)中,分子筛的干燥处理没有特别的限定,例如可以按照常规方法在120℃下干燥6小时。在本发明中,所述晶化后的母液可以按照本领域常规的方法制备,例如,当所述十元环硅铝分子筛为zsm-22分子筛时,晶化后的母液的制备方法可以参照文献o.murazaetal.,microporousandmesoporousmaterials206(2015)136–143;当所述十元环硅铝分子筛为zsm-48分子筛时,晶化后的母液的制备方法可以参照文献p.me′riaudeauetal/journalofcatalysis,1999(185),435–444,或者参照美国专利申请us5961951。在一种实施方式中,所述十元环硅铝分子筛的晶化母液的制备过程包括:制备含硅溶液、含铝溶液、碱性液体,将上述液体混合进行成胶,随后在一定温度下进行晶化。在本发明中,所述十元环硅铝分子筛的类型没有特别的限定,例如可以为zsm-11、zsm-22、zsm-23、zsm-35、zsm-48、zsm-57、nu-10、nu-13、nu-87、eu-1、eu-13和itq-13中的至少一种。在优选情况下,所述十元环硅铝分子筛为zsm-22、zsm-23和zsm-48中的至少一种,更优选为zsm-22和/或zsm-48,最优选为zsm-22。在本发明所述的加氢异构化催化剂中,优选地,所述活性金属组分在分子筛上呈高度分散状态,具体地,所述活性金属组分的单个颗粒的尺寸小于3nm,例如可以为0.1-2.8nm。在本发明所述的加氢异构化催化剂中,所述活性金属组分为viii族贵金属中的至少两种。viii族贵金属为钌、锇、钯、铂、铑和铱。优选情况下,所述活性金属组分为铂组分和钯组分的组合。进一步优选地,pt组分与pd组分的摩尔比为1:2-10,更优选为1:2-4。在本发明中,所述活性金属组分可以由活性金属组分前驱体提供。所述活性金属组分前驱体选自含viii族贵金属元素的化合物。所述含viii族贵金属元素的化合物可以选自含viii族贵金属元素的硝酸盐、氯化物、硫酸盐、甲酸盐、乙酸盐、磷酸盐、柠檬酸盐、草酸盐、碳酸盐、碱式碳酸盐、氢氧化物、磷酸盐、磷化物、硫化物、铝酸盐、钼酸盐、钨酸盐和水溶性氧化物中的一种或多种。在本发明所述的加氢异构化催化剂中,以催化剂的总重量为基准,以元素计的活性金属组分的含量为0.1-5重量%,优选为0.2-3重量%,更优选为0.4-1重量%。本发明还提供了制备上述加氢异构化催化剂的方法,该方法包括以下步骤:(i)采用浸渍法将活性金属组分前驱体和有机络合剂负载在载体上,然后进行干燥、焙烧,得到半成品催化剂;(ii)以含有有机络合剂的溶液作为浸渍液,对步骤(i)所得半成品催化剂进行浸渍,然后进行干燥。在本发明中,通过两步浸渍法制备所述加氢异构化催化剂,第一步浸渍和第二步浸渍分别用于引入活性金属组分和有机络合剂,在第一步浸渍过程中加入有机络合剂并使之通过焙烧转化为炭,不仅能够提高催化剂的活性,而且能够有效地长时间保持催化剂的高活性,从而大大提高催化剂的使用寿命。推测其原因可能是因为第一步浸渍过程中加入的有机络合剂,有机络合剂的存在阻碍了焙烧过程中活性金属在氧化铝上的聚集,使其在分子筛上分散更加均匀;同时,第一步浸渍后焙烧能够使金属化合物转化为金属氧化物,使有机络合剂转化为炭,从而使活性金属与载体之间的结合更加牢固,提高了催化剂的活性和稳定性。而在第二步浸渍过程中加入的有机络合剂覆盖在催化剂表面,能够有效防止活性金属之间在还原过程中的聚集,保证高金属分散度,从而进一步提高了催化剂的活性。因此,按照本发明所述的方法可以有效解决活性金属组分在催化剂上分布不均匀的问题。在本发明所述的方法中,步骤(i)所用有机络合剂可以选自含氧有机物、有机酸和含氮有机物中的至少一种。所述含氧有机物可以为二元以上的多元醇,更进一步优选为碳原子数为2-6的多元醇或其低聚体或多聚体,如乙二醇、丙三醇、聚乙二醇、二乙二醇、丁二醇中的一种或多种。所述聚乙二醇的分子量优选为200-1500。所述有机酸优选为c2-c7的含一个或多个羧基的化合物,具体可以为乙酸、马来酸、草酸、氨基三乙酸、1,2-环己烷二胺四乙酸、柠檬酸、酒石酸和苹果酸中的一种或多种。所述含氮有机物优选选自有机胺和有机铵盐中的一种或多种。所述有机胺优选为c2-c7的含一个或多个氨基的化合物,可以是伯胺、仲胺或叔胺,特别优选为乙二胺。所述有机铵盐优选为edta。优选地,步骤(1)中所述有机络合剂选自有机酸中的一种或多种,更优选地,步骤(1)所述有机络合剂选自c2-c7的脂肪酸中的一种或多种。使用有机酸作为步骤(1)的有机络合剂,可以获得具有更高活性的加氢异构化催化剂。在步骤(i)中,所述有机络合剂与所述活性金属组分前驱体的摩尔比可以为2-100:1,优选为10-50:1。在步骤(i)中,浸渍时对所述浸渍的温度没有特别限定,可以是浸渍液所能达到的各种温度,对浸渍的时间没有特别限定,只要能负载上所需量的所需组分即可,例如:浸渍的温度可以为15-60℃,浸渍时间可以为0.5-5小时。浸渍时含有机络合剂液体与载体的质量比可以为0.6:1~2:1,优选为0.8:1~1.4:1。在步骤(i)中,所述干燥的条件没有特别的限定,可以是本领域常用的各种干燥条件,优选地,步骤(1)所述干燥的温度为100-250℃,时间为1-12小时。在步骤(i)中,优选地,所述焙烧的条件使得以半成品催化剂的总量为基准,半成品催化剂中炭含量为0.05-0.5重量%,优选为0.1-0.4重量%。在本发明中,可以通过控制焙烧条件中的焙烧温度和可燃性气体的通入量来获得上述炭含量,所述可燃性气体可以为各种氧气含量不低于20体积%的气体,如空气、氧气以及它们的混合气体中的一种或多种。所述可燃性气体的通入量不低于0.2升/克·小时。所述可燃性气体的通入,一方面满足燃烧的条件,使得活性金属组分的盐转化为氧化物,使有机络合剂转化为炭;另一方面也能将燃烧形成的二氧化碳和水以及其他成分排放出去,以避免沉积在催化剂上造成对活性相的空位阻碍。优选情况下,可燃性气体的通入量为0.2-20升/(克·小时),优选为0.3-10升/(克·小时)。此处的“克”表示载体的重量。在步骤(i)中,所述焙烧的温度可以为350-500℃,优选为360-450℃,焙烧的时间可以为0.5-8小时,优选为1-6小时。控制焙烧温度在上述范围内即可保证有机络合剂能以上述含量范围将炭形成于载体上,得到半成品催化剂。在步骤(ii)中,所用有机络合剂的选择范围与步骤(i)相同。步骤(ii)中所用有机络合剂可以与步骤(i)所用有机络合剂相同或不同。步骤(ii)所用有机络合剂与活性金属的摩尔比可以与步骤(i)相同。步骤(ii)所用浸渍、干燥条件与步骤(i)相同。在本发明所述的方法中,步骤(ii)干燥后得到的催化剂不需要再进行焙烧。根据本发明,所述加氢异构化催化剂的制备方法还可以包括对催化剂进行还原处理。本发明对还原条件不进行限定。一般的,还原气氛为氢气,还原温度可以为300-500℃,还原时间可以为2-4小时。本发明还提供了一种加氢裂化尾油的加氢处理方法,该方法包括:在加氢异构化反应条件下,将加氢裂化尾油与催化剂接触反应,其中,所述催化剂为本发明提供的所述加氢异构化催化剂或者根据上述方法制备的加氢异构化催化剂。在本发明中,所述加氢裂化尾油的馏程一般可以为350-500℃(采用模拟蒸馏方法在常压下测定)。在本发明中,所述加氢异构化反应条件没有特别的限定,只要足以使加氢裂化尾油发生加氢异构化反应即可。一般地,所述加氢异构化反应条件可以包括:温度为200-500℃,优选为250-400℃,更优选为300-350℃;压力为1-30mpa,优选为2-20mpa,更优选为5-20mpa,本文中所述的压力是指绝对压力;空速为0.1-5h-1,优选为0.1-3h-1,更优选为0.5-2h-1;氢油体积比为50-3000,优选为300-3000,更优选为400-600。按照本发明所述的方法使加氢裂化尾油与加氢异构化催化剂接触,进行加氢异构化反应,能够获得较高的异构化产物收率;并且,异构化产物在具有较高的黏度指数的同时,还具有较低的倾点,适于作为润滑油基础油。以下结合实施例详细说明本发明,但并不因此限制本发明的范围。以下实施例和对比例中,采用商购自日本理学电机工业株式会社的3271e型x射线荧光光谱仪,对测定样品中各元素的含量进行分析测定,样品测试前在600℃下焙烧3小时。以下实施例和对比例中采用美国micromeritics公司digisorb2500型自动吸附仪测定样品的比表面积、外表面积,样品测试前在600℃下焙烧3小时,测量方法均可以按照astmd4222-98标准方法进行。以下实施例和对比例中,干基是指一定量的物料在空气气氛下于马弗炉中在600℃焙烧4小时后得到的产物的重量与焙烧前物料的重量之比的百分数。即干基=(焙烧后得到的产物的重量÷焙烧前物料的重量)×100%。以下实施例和对比例中催化剂半成品中炭含量使用日本horiba公司生产的emia-320v碳硫分析仪进行分析测定。以下实施例和对比例中黏度指数是按照gb/t1995-1998方法测量,倾点是按照gb/t3535方法测量。制备例1(1)制备晶化后的母液取36.3克含40重量%的sio2的硅溶胶,1.77克分析纯的al2(so4)3·18h2o,3.94克分析纯的koh以及8.44克己二胺待用。将己二胺与硅溶胶混合,将koh和al2(so4)3·18h2o以及89.4克去离子水混合,随后将两种溶液混合,搅拌1h后移入反应釜中,在160℃下晶化72小时。(2)制备滤饼将工序(1)中制备的晶化后的母液进行过滤,以滤饼上没有滤液计时,继续抽滤5分钟,获得的滤饼f-1,该滤饼f-1的干基含量为11.2%,氧化硅/氧化铝摩尔比为30.2。(3)制备分子筛前体将滤饼f-1以25℃/分钟的升温速率从室温升到450℃,恒温4小时。升温过程中焙烧炉为密闭焙烧炉,获得分子筛前体c-1,其27alnmr图谱见图1。(4)制备分子筛成品将分子筛前体c-1放入浓度为1m的hcl溶液中进行密闭水热处理。其中,液固比为50,水热处理的温度为180℃,水热处理的时间为3小时,水热处理结束后将产物过滤、水洗,直至滤液ph为7,经120℃干燥4小时后,在550℃下焙烧4小时,获得的zsm-22分子筛成品h-1。该分子筛的xrd图谱、27alnmr图谱以及氮气吸附-脱附曲线分别见图2、图3和图4。由图4可以看出,本发明所述的zsm-22分子筛在低温氮气吸附-脱附曲线p/p0=0.4-0.99处出现一个闭合滞后环,且所述闭合滞后环的起始位置在p/p0=0.4-0.5处。制备对比例1根据制备例1的方法制备zsm-22分子筛,所不同的是,在步骤(2)中,以滤饼上没有滤液计时,继续抽滤50分钟,获得的滤饼df-1,该滤饼df-1的干基含量为46.5%。制得zsm-22分子筛成品dh-1。制备例2根据制备例1的方法制备zsm-22分子筛,所不同的是,在步骤(3)中,将滤饼f-1以5℃/分钟的升温速率从室温升到350℃,恒温14小时。升温过程中焙烧炉为密闭焙烧炉,获得分子筛前体c-2。制得zsm-22分子筛成品h-2。制备例3根据制备例1的方法制备zsm-22分子筛,所不同的是,在步骤(3)中,将滤饼f-1以15℃/分钟的升温速率从室温升到850℃,恒温4小时。升温过程中通入空气,空气流速为1.0升/分钟,获得分子筛前体c-3。制得zsm-22分子筛成品h-3。制备例4根据制备例1的方法制备zsm-22分子筛,所不同的是,在步骤(4)中,将分子筛前体c-1放入浓度为1.0m的柠檬酸溶液中进行密闭水热处理。其中,液固比为100,水热处理的温度为180℃,水热处理的时间为2小时,水热处理结束后将产物过滤、水洗,直至滤液ph值为7,经120℃干燥4小时后,在550℃下焙烧4小时,获得的zsm-22分子筛成品h-4。制备例5根据制备例1的方法制备zsm-22分子筛,所不同的是,在步骤(4)中,将分子筛前体c-1放入浓度为0.05m的柠檬酸溶液中进行密闭水热处理。其中,液固比为10,水热处理的温度为90℃,水热处理的时间为0.1小时,水热处理结束后将产物过滤、水洗,直至滤液ph值为7,经120℃干燥4小时后,在550℃下焙烧4小时,获得的zsm-22分子筛成品h-5。制备例6根据制备例1的方法制备zsm-22分子筛,所不同的是,在步骤(4)中,将分子筛前体c-1放入浓度为1m的盐酸溶液中进行密闭水热处理。其中,液固比为50,水热处理的温度为180℃,水热处理的时间为3小时,水热处理结束后将产物过滤、水洗,直至滤液ph值为4,经120℃干燥4小时后,在550℃下焙烧4小时,获得的zsm-22分子筛成品h-6。制备对比例2根据制备例1中工序(1)制备晶化后的母液,接着过滤,并将过滤后的得到的滤饼在120℃下干燥4小时,然后在550℃下焙烧4小时,获得分子筛前体dc-2。将分子筛前体dc-2与10倍体积的0.5m盐酸溶液在90℃下进行铵交换处理4小时,经过滤后重新与10倍体积的0.5m盐酸溶液在90℃下进行铵交换处理4小时,最后经过滤、干燥和550℃下焙烧4小时后获得zsm-22分子筛成品dh-2。制备对比例3根据制备例1中工序(1)制备晶化后的母液,接着过滤,并将过滤后的得到的滤饼在120℃下干燥4小时,然后在850℃下焙烧4小时,获得分子筛前体dc-3。将分子筛前体dc-3与10倍体积的0.5m盐酸溶液在90℃下进行铵交换处理4小时,经过滤后重新与10倍体积的0.5m盐酸溶液在90℃下进行铵交换处理4小时,最后经过滤、干燥和550℃下焙烧4小时后获得zsm-22分子筛成品dh-3,其27alnmr图谱如图5所示,氮气吸附-脱附曲线如图6所示。测试例1(1)采用美国micromeritics公司digisorb2500型自动吸附仪测定上述制备例和制备对比例制备的分子筛成品的介孔面积、比表面积,并计算介孔面积占比表面积的比例,结果如下表1所示。(2)采用商购自日本理学电机工业株式会社的3271e型x射线荧光光谱仪,对上述制备例和制备对比例制备的分子筛前体和分子筛成品中各元素的含量进行分析测定,确定硅铝比和五配位铝的含量,结果如下表1所示。表1实施例1将制备例1制备的40g分子筛h-1与40g氧化铝进行混合、挤条、干燥,得到载体e-1。将0.4克二氯四氨合铂、0.6克二氯四按钯以及3.2克柠檬酸倒入100克去离子水中,搅拌至均匀。将80克载体e-1倒入上述溶液中,在室温下浸渍4小时。随后,将上述催化剂前体在120℃下干燥4小时。接着将其在通入空气流的状态下进行焙烧,焙烧温度为450℃,时间为4小时,气剂比为2.0升/(克·小时),得到半成品催化剂。将该半成品催化剂再次放入含有3.2克柠檬酸的100克去离子水溶液。浸渍4小时后,在120℃下干燥4小时,获得的催化剂ic-1。实施例2-6和对比例1-3根据实施例1的方法制备催化剂,所不同的是,分别用制备例2-6制备的分子筛h-2至h-6以及制备对比例1-3制备的分子筛dh-1至dh-3代替实施例1中使用的分子筛h-1,从而制得催化剂ic-2至ic-6以及参比催化剂dic-1至dic-3。实施例7根据实施例1的方法制备载体e-1。将0.4克二氯四氨合铂、0.6克二氯四按钯以及16克柠檬酸倒入100克去离子水中,搅拌至均匀。将80克载体e-1倒入上述溶液中,在室温下浸渍4小时。随后,将上述催化剂前体在120℃下干燥4小时。接着将其在通入空气流的状态下进行焙烧,焙烧温度为450℃,时间为4小时,气剂比为2.0升/(克·小时),得到半成品催化剂。将该半成品催化剂再次放入含有16克柠檬酸的100克去离子水溶液。浸渍4小时后,在120℃下干燥4小时,获得的催化剂ic-7。实施例8根据实施例1的方法制备载体e-1。将0.4克二氯四氨合铂、0.6克二氯四按钯以及18克edta倒入100克去离子水中,搅拌至均匀。将80克载体e-1倒入上述溶液中,在室温下浸渍4小时。随后,将上述催化剂前体在120℃下干燥4小时。接着将其在通入空气流的状态下进行焙烧,焙烧温度为450℃,时间为4小时,气剂比为2.0升/(克·小时),得到半成品催化剂。将该半成品催化剂再次放入含有6.4克二甘醇的100克去离子水溶液。浸渍4小时后,在120℃下干燥4小时,获得的催化剂ic-8。实施例9根据实施例1的方法制备载体e-1。将0.4克二氯四氨合铂、0.6克二氯四按钯以及20克丁二醇倒入100克去离子水中,搅拌至均匀。将80克载体e-1倒入上述溶液中,在室温下浸渍4小时。随后,将上述催化剂前体在120℃下干燥4小时。接着将其在通入空气流的状态下进行焙烧,焙烧温度为350℃,时间为4小时,气剂比为1.0升/(克·小时),得到半成品催化剂。将该半成品催化剂再次放入含有3.2克柠檬酸的100克去离子水溶液。浸渍4小时后,在120℃下干燥4小时,获得的催化剂ic-9。实施例10根据实施例1的方法制备载体e-1。将0.4克二氯四氨合铂、0.6克二氯四按钯以及19克乙二胺倒入100克去离子水中,搅拌至均匀。将80克载体e-1倒入上述溶液中,在室温下浸渍4小时。随后,将上述催化剂前体在120℃下干燥4小时。接着将其在通入空气流的状态下进行焙烧,焙烧温度为350℃,时间为4小时,气剂比为1.0升/(克·小时),得到半成品催化剂。将该半成品催化剂再次放入含有1.0克柠檬酸的100克去离子水溶液。浸渍4小时后,在120℃下干燥4小时,获得的催化剂ic-10。测试例2(1)以下实施例和对比例中催化剂半成品中炭含量使用日本horiba公司生产的emia-320v碳硫分析仪进行分析测定,结果如下表3所示。(2)分别将100g、20-30目上述实施例和对比例制备的催化剂放入反应管中,在氢气气氛下还原4小时,还原温度为400℃,还原时氢气压力为常压。还原结束后降温到120℃,进入加氢裂化尾油,反应温度为310℃,油料的体积空速为1.0h-1,调整氢气压力为10.0mpa,调整氢气流量使氢油体积比为500。加氢裂化尾油性质如下表2所示,催化剂评价结果如下表3所示。表2分析项目分析数据分析方法20℃密度/(kg/m3)843.6sh/t0604-2000运动黏度/(mm2/s)80℃7.021gb/t265-88100℃4.664gb/t265-88倾点/℃+42氮质量分数/(μg/g)<1硫质量分数/(μg/g)3sh/t0842-2010表3由上表3的数据可以看出,采用本发明所述的加氢异构化催化剂对加氢裂化尾油进行加氢处理,所获得的目标产物的黏度指数较高、倾点较低且收率较高。以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1