一种取代的磷酰胺类化合物的制备方法与流程
本发明涉及有机合成方法学技术领域,尤其涉及一种取代的磷酰胺类化合物的制备方法。
背景技术:
磷酰胺类化合物作为药物和有机材料,有着广泛的生物活性和应用价值。其结构中n-p键的形成是其合成中的关键步骤。
n-p键在许多生物活性化合物和有机材料中均普遍存在,因此n-p键的形成,已成为许多化合物合成中的关键。在过去的几十年中,使用磷酰氯、磷酸与胺反应和过渡金属催化下构建n-p键已成为一个传统的主流方法。然而,这些传统方法反应条件剧烈,对水分和氧气敏感,并且原子经济性低。随着绿色环保和原子经济性概念的深入发展,如何在更加温和的条件下构建n-p键成为亟待解决的问题。
技术实现要素:
有鉴于此,本发明要解决的技术问题在于提供一种取代的磷酰胺类化合物的制备方法,一步反应即可,操作简单,且条件温和。
本发明提供了一种取代的磷酰胺类化合物的制备方法,包括以下步骤:
将电解质、取代的胺、取代的膦氧化物和溶剂分别加入到电解池中,通电搅拌反应,得到式ⅰ所示的取代的磷酰胺类化合物;
其中,r1和r2独立的选自:卤素,取代或非取代的c1~c5的烷氧基、c1~c5的烷基、c6~c18的芳基或c6~c18的杂芳基;
r3和r4独立的选自:取代或非取代的c4~c12的烷基、c4~c12的烷氧基、c4~c12的环烷基、c6~c12的芳基或c6~c12的杂芳基;
或者r3、r4和相连的n原子共同形成取代或非取代的c3~c12的环烷基、杂环基、芳基或杂芳基。
本发明采用的电解质优选为卤盐电解质,即含卤素原子的盐,如碘化铵、碘化钾、四丁基碘化铵、溴化钾和氯化钾等卤盐电解质,本发明选用的卤盐电解质可以是以上任意一种,或几种的组合。
所述取代的胺结构如下:
其中,r3和r4范围同上,在此不再赘述。
在本发明的某些具体实施例中,所述r3、r4和相连的n原子共同形成吗啉基、哌啶基、哌嗪基、吡啶基、吡咯基、吡嗪基等c3~c12的杂环基团,上述杂环基团可以带有c1~c5烷基、c1~c5烷氧基、羟基、卤素、硝基、氰基、磺酰基、羟基烷基、苯氧基、苯基羰基、苄基等取代基,还可以和其他脂肪环或芳环稠合成为桥环基团或螺环基团。
所述取代的膦氧化物结构如下:
其中,r1和r2范围同上,在此不再赘述。
在本发明的某些具体实施例中,所述膦氧化物可以为二烷基膦氧,如二甲基氧化膦或氧化二乙基膦等;或者二芳基膦氧,如二苯基磷氧,双(对甲基苯基)氧化膦,双(3,5-二甲基苯基)氧化膦,双(对甲氧基苯基)氧化膦,双(3-甲氧基苯基)氧化膦,双(3,5-二甲氧基苯基)氧化膦,双(对氯苯基)氧化膦,双(对氟苯基)氧化膦,2,2'-双-(2-萘基)氧化膦或苯基乙氧基氧化膦等。
上述电解质、取代的胺、取代的膦氧化物的摩尔比优选为(3~4):(2~4):1。
反应过程中,所述取代的膦氧化物的起始浓度优选为0.03~0.15mol/l-1。
所述反应的溶剂优选为甲醇和/或乙醇。
所述通电的电流密度优选为8~20ma/cm-2,电极采用铂电极和石墨电极。
所述反应的温度优选为15~40℃。
反应后还包括分离提纯。
所述分离提纯包括柱层析色谱、液相色谱、蒸馏和重结晶中的任意一种或几种的组合。
更优选地,所述分离提纯方式为柱层色谱。
优选地,所述柱层色谱的洗脱剂为乙酸乙酯/甲醇,但并不局限于该种洗脱剂,只要符合洗脱目的的试剂均可以使用。
上述反应的反应式如下:
通过本发明提供的方法,可以一步构建取代的磷酰胺类化合物,由此可以衍生出许多重要的具有生理活性的中间体和有机材料。
在本发明的某些具体实施例中,所制备的取代的磷酰胺类化合物具有以下3aa~3ck任一结构:
上述结构式中的单键表示甲基。
与现有技术相比,本发明提供了一种取代的磷酰胺类化合物的制备方法,包括以下步骤:将电解质、取代的胺、取代的膦氧化物和溶剂分别加入到电解池中,通电搅拌反应,得到式ⅰ所示的取代的磷酰胺类化合物。本发明首次实现了在通电条件下,取代的膦氧化物与胺反应,得到取代的磷酰胺类化合物。该方法绿色高效,一步反应即可得到产品,操作简单,且条件温和。
附图说明
图1为实施例1提供的产物1h核磁共振谱图;
图2为实施例1提供的产物13c核磁共振谱图;
图3为实施例1提供的产物31p核磁共振谱图;
图4为实施例2提供的产物1h核磁共振谱图;
图5为实施例2提供的产物13c核磁共振谱图;
图6为实施例2提供的产物31p核磁共振谱图;
图7为实施例3提供的产物1h核磁共振谱图;
图8为实施例3提供的产物13c核磁共振谱图;
图9为实施例3提供的产物31p核磁共振谱图;
图10为实施例4提供的产物1h核磁共振谱图;
图11为实施例4提供的产物13c核磁共振谱图;
图12为实施例4提供的产物31p核磁共振谱图;
图13为实施例5提供的产物1h核磁共振谱图;
图14为实施例5提供的产物13c核磁共振谱图;
图15为实施例5提供的产物31p核磁共振谱图;
图16为实施例6提供的产物1h核磁共振谱图;
图17为实施例6提供的产物13c核磁共振谱图;
图18为实施例6提供的产物31p核磁共振谱图;
图19为实施例7提供的产物1h核磁共振谱图;
图20为实施例7提供的产物13c核磁共振谱图;
图21为实施例7提供的产物31p核磁共振谱图;
图22为实施例8提供的产物1h核磁共振谱图;
图23为实施例8提供的产物13c核磁共振谱图;
图24为实施例8提供的产物31p核磁共振谱图;
图25为实施例9提供的产物1h核磁共振谱图;
图26为实施例9提供的产物13c核磁共振谱图;
图27为实施例9提供的产物31p核磁共振谱图;
图28为实施例10提供的产物1h核磁共振谱图;
图29为实施例10提供的产物13c核磁共振谱图;
图30为实施例10提供的产物31p核磁共振谱图。
具体实施方式
为了进一步说明本发明,下面结合实施例对本发明提供的取代的磷酰胺类化合物的制备方法进行详细描述。
以下实施例中所使用的部分取代的膦氧化物根据文献方法合成,其余部分的取代的膦氧化物、所有胺及试剂均为直接购买的分析纯试剂,使用前未经其他处理,所用溶剂或洗脱剂购于国药。
实施例1
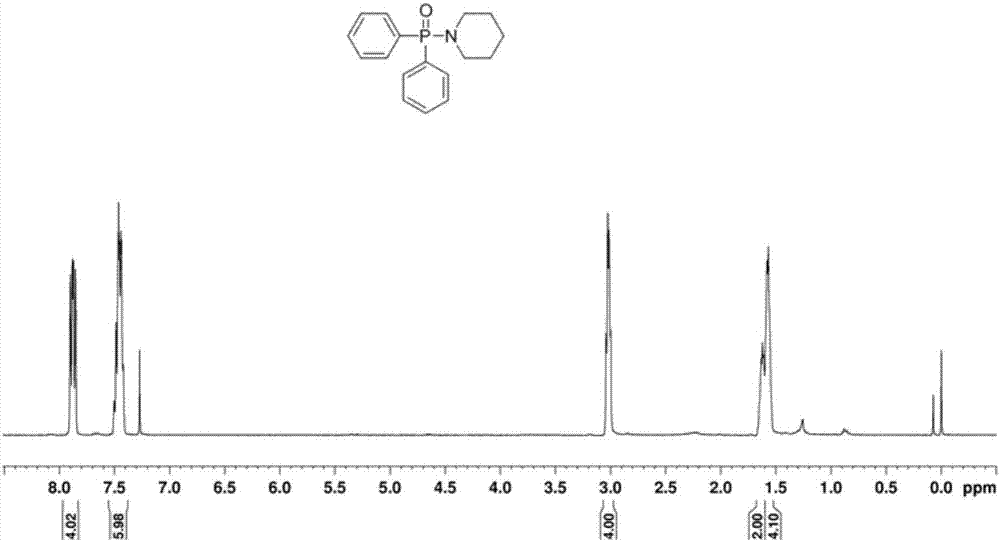
在15ml电解池中加入二苯基膦氧(0.3mmol,60.6mg)、哌啶(0.9mmol,76.5mg)、碘化钾(1mmol,166mg)和乙醇(10ml),恒电流30ma条件下搅拌反应。反应完成后(tlc跟踪检测),旋干得到的残留物用乙酸乙酯/甲醇体系作为洗脱剂过色谱柱得到产物3ab,产率为75%,纯度>99%。
通过核磁共振波谱仪对所述加成产物进行分析,结果参见图1~图3。其中,图1为本发明实施例1提供的产物1h核磁共振(1hnmr)谱图;图2为本发明实施例1提供的产物13c核磁共振(13cnmr)谱图;图3为本发明实施例1提供的产物31p核磁共振(31pnmr)谱图。由图1~图3可知,本发明实施例1提供的产物的结构如3ab所示。
表征数据:
1hnmr(400mhz,cdcl3,ppm):δ=7.90–7.85(m,4h),7.50–7.42(m,6h),3.04-3.00(m,4h),1.63-1.61(m,2h),1.58-1.57(m,4h)。
13cnmr(100mhz,cdcl3,ppm):δ=131.3(d,j=9.7hz),130.9(d,j=128.7hz),130.6(d,j=2.7hz),127.5(d,j=12.3hz),44.7,25.2(d,j=6.8hz),23.5。
31pnmr(162mhz,cdcl3,ppm)δ=28.99.hrms(esi)calcdforc17h21nop[m+h]+286.1361,found286.1361。
实施例2
在15ml电解池中加入二苯基膦氧(0.3mmol,60.6mg)、4-羟基哌啶(0.9mmol,90.9mg)、碘化钾(1mmol,166mg)和乙醇(10ml),恒电流30ma条件下搅拌反应。反应完成后(tlc跟踪检测),旋干得到的残留物用乙酸乙酯/甲醇体系作为洗脱剂过色谱柱得到产物3ae,产率为83%,纯度98%。
通过核磁共振波谱仪对所述加成产物进行分析,结果参见图4~图6。其中,图4为本发明实施例2提供的产物1h核磁共振(1hnmr)谱图;图5为本发明实施例2提供的产物13c核磁共振(13cnmr)谱图;图6为本发明实施例2提供的产物31p核磁共振(31pnmr)谱图。由图4~图6可知,本发明实施例2提供的产物的结构如3ae所示。
对所述产物进行测定,其表征数据为:
1hnmr(400mhz,cdcl3,ppm):δ=7.87–7.82(m,4h),7.50–7.43(m,6h),3.83-3.79(m,1h),3.48(s,1h),3.31-3.28(m,2h),2.87-2.80(m,2h),1.91-1.88(m,2h),1.62-1.53(m,2h)。
13cnmr(100mhz,cdcl3,ppm):δ=132.1(d,j=9.1hz),131.7(d,j=2.4hz),131.3(d,j=129.4hz),128.6(d,j=12.4hz),67.2,42.8,34.8(d,j=6.5hz)。
31pnmr(162mhz,cdcl3,ppm)δ=29.77.hrms(esi)calcdforc17h21no2p[m+h]+302.1310,found302.1309。
实施例3
在15ml电解池中加入二苯基膦氧(0.3mmol,60.6mg)、1-甲磺酰基哌嗪(0.9mmol,147.6mg)、碘化钾(1mmol,166mg)和乙醇(10ml),恒电流30ma条件下搅拌反应。反应完成后(tlc跟踪检测),旋干得到的残留物用乙酸乙酯/甲醇体系作为洗脱剂过色谱柱得到产物3ah,产率为54%,纯度>99%。
通过核磁共振波谱仪对所述加成产物进行分析,结果参见图7~9。其中,图7为本发明实施例3提供的产物1h核磁共振(1hnmr)谱图;图8为本发明实施例3提供的产物13c核磁共振(13cnmr)谱图;图9为本发明实施例3提供的产物31p核磁共振(31pnmr)谱图。由图7~9可知,本发明实施例3提供的产物结构如3ah所示。
对所述产物进行测定,其表征数据为:
1hnmr(400mhz,cdcl3,ppm):δ=7.88–7.84(m,4h),7.56–7.48(m,6h),3.25-3.22(m,8h),2.80(s,3h)。
13cnmr(100mhz,cdcl3,ppm):δ=132.19(d,j=9.1hz),132.18(d,j=2.7hz),130.5(d,j=128.7hz),128.8(d,j=12.5hz),46.2(d,j=4.3hz),44.9,34.3。
31pnmr(162mhz,cdcl3,ppm)δ=30.23.hrms(esi)calcdforc17h22n2o3ps[m+h]+365.1079,found365.1082。
实施例4
在15ml电解池中加入二苯基膦氧(0.3mmol,60.6mg)、3-吡咯啉(0.9mmol,62.1mg)、碘化钾(1mmol,166mg)和乙醇(10ml),恒电流30ma条件下搅拌反应。反应完成后(tlc跟踪检测),旋干得到的残留物用乙酸乙酯/甲醇体系作为洗脱剂过色谱柱得到产物3ak,产率为80%,纯度>99%。
通过核磁共振波谱仪对所述加成产物进行分析,结果参见图10~12。其中,图10为本发明实施例4提供的产物1h核磁共振(1hnmr)谱图;图11为本发明实施例4提供的产物13c核磁共振(13cnmr)谱图;图12为本发明实施例4提供的产物31p核磁共振(31pnmr)谱图。由图10~12可知,本发明实施例4提供的产物结构如3ak所示。
对所述产物进行测定,其表征数据为:
1hnmr(400mhz,cdcl3,ppm):δ=7.93–7.88(m,4h),7.52–7.44(m,6h),5.77(s,2h),3.99(d,j=5.8hz,4h)。
13cnmr(100mhz,cdcl3,ppm):δ=132.2(d,j=9.4hz),132.1(d,j=128.7hz),131.7(d,j=2.5hz),128.6(d,j=12.4hz),126.5(d,j=7.3hz),54.2(d,j=3.5hz)。
31pnmr(162mhz,cdcl3,ppm)δ=25.06.hrms(esi)calcdforc16h17nop[m+h]+270.1048,found270.1048。
实施例5
在15ml电解池中加入二苯基膦氧(0.3mmol,60.6mg)、二乙胺(0.9mmol,65.7mg)、碘化钾(1mmol,166mg)和乙醇(10ml),恒电流30ma条件下搅拌反应。反应完成后(tlc跟踪检测),旋干得到的残留物用乙酸乙酯/甲醇体系作为洗脱剂过色谱柱得到产物3al,产率为57%,纯度>99%。
通过核磁共振波谱仪对所述加成产物进行分析,结果参见图13~15。其中,图13为本发明实施例5提供的产物1h核磁共振(1hnmr)谱图;图14为本发明实施例5提供的产物13c核磁共振(13cnmr)谱图;图15为本发明实施例5提供的产物31p核磁共振(31pnmr)谱图。由图13~15可知,本发明实施例5提供的产物结构如3al所示。
对所述产物进行测定,其表征数据为:
1hnmr(400mhz,cdcl3,ppm):δ=7.88–7.84(m,4h),7.50–7.42(m,6h),3.07(dt,j=17.9,7.0hz,4h),1.10(t,j=7.0hz,6h)。
13cnmr(100mhz,cdcl3,ppm):δ=132.5(d,j=127.8hz),132.2(d,j=9.1hz),131.5(d,j=2.7hz),128.3(d,j=12.3hz),39.2(d,j=3.4hz),14.0(d,j=3.9hz)。
31pnmr(162mhz,cdcl3,ppm)δ=30.56.hrms(esi)calcdforc16h21nop[m+h]+274.1361,found274.1360。
实施例6
在15ml电解池中加入二苯基膦氧(0.3mmol,60.6mg)、n-甲基苄胺(0.9mmol,108.9mg)、碘化钾(1mmol,166mg)和乙醇(10ml),恒电流30ma条件下搅拌反应。反应完成后(tlc跟踪检测),旋干得到的残留物用乙酸乙酯/甲醇体系作为洗脱剂过色谱柱得到产物3am,产率为77%,纯度>99%。
通过核磁共振波谱仪对所述加成产物进行分析,结果参见图16~18。其中,图16为本发明实施例6提供的产物1h核磁共振(1hnmr)谱图;图17为本发明实施例6提供的产物13c核磁共振(13cnmr)谱图;图18为本发明实施例6提供的产物31p核磁共振(31pnmr)谱图。由图16~18可知,本发明实施例6提供的产物结构如3am所示。
对所述产物进行测定,其表征数据为:
1hnmr(400mhz,cdcl3,ppm):δ=7.96–7.91(m,4h),7.51–7.42(m,6h),7.37–7.32(m,4h),7.28–7.24(m,1h),4.13(d,j=7.7hz,2h),2.54(d,j=11.0hz,3h)。
13cnmr(100mhz,cdcl3,ppm):δ=137.4(d,j=7.1hz),132.2(d,j=9.2hz),131.7(d,j=2.7hz),131.6(d,j=128.0hz),128.6,128.4(d,j=4.5hz),127.9,127.2,52.8(d,j=3.0hz),33.6(d,j=2.6hz)。
31pnmr(162mhz,cdcl3,ppm)δ=31.66.hrms(esi)calcdforc20h21nop[m+h]+322.1361,found322.1359。
实施例7
在15ml电解池中加入双(4-甲基苯基)氧化膦(0.3mmol,68.7mg)、吗啡啉(0.9mmol,78.3mg)、碘化钾(1mmol,166mg)和乙醇(10ml),恒电流30ma条件下搅拌反应。反应完成后(tlc跟踪检测),旋干得到的残留物用乙酸乙酯/甲醇体系作为洗脱剂过色谱柱得到产物3ba,产率为77%,纯度99%。
通过核磁共振波谱仪对所述加成产物进行分析,结果参见图19~21。其中,图19为本发明实施例7提供的产物1h核磁共振(1hnmr)谱图;图20为本发明实施例7提供的产物13c核磁共振(13cnmr)谱图;图21为本发明实施例7提供的产物31p核磁共振(31pnmr)谱图。由图19~21可知,本发明实施例7提供的产物的结构为3ba所示的结构。
对所述产物进行测定,其表征数据为:
1hnmr(400mhz,cdcl3,ppm):δ=7.77–7.72(m,4h),7.27–7.26(m,4h),3.70(s,4h),3.06(s,4h),2.37(s,6h)。
13cnmr(100mhz,cdcl3,ppm):δ=142.3(d,j=2.6hz),132.3(d,j=9.4hz),129.4(d,j=12.7hz),127.6(d,j=131.0hz),67.2(d,j=6.7hz),44.9,21.5。
31pnmr(162mhz,cdcl3,ppm)δ=29.89.hrms(esi)calcdforc18h23no2p[m+h]+316.1466,found316.1464。
实施例8
在15ml电解池中加入双(4-甲氧基苯基)氧化膦(0.3mmol,78.3mg)、吗啡啉(0.9mmol,78.3mg)、碘化钾(1mmol,166mg)和乙醇(10ml),恒电流30ma条件下搅拌反应。反应完成后(tlc跟踪检测),旋干得到的残留物用乙酸乙酯/甲醇体系作为洗脱剂过色谱柱得到产物3da,产率为76%,纯度98%。
通过核磁共振波谱仪对所述加成产物进行分析,结果参见图22~24。其中,图22为本发明实施例8提供的产物1h核磁共振(1hnmr)谱图;图23为本发明实施例8提供的产物13c核磁共振(13cnmr)谱图;图24为本发明实施例8提供的产物31p核磁共振(31pnmr)谱图。由图22~24可知,本发明实施例8提供的产物结构如3da所示。
对所述产物进行测定,其表征数据为:
1hnmr(400mhz,cdcl3,ppm):δ=7.80–7.75(m,4h),6.98–6.96(m,4h),3.83(s,6h),3.69(t,j=4.4hz,4h),3.07-3.03(m,4h)。
13cnmr(100mhz,cdcl3,ppm):δ=162.4(d,j=2.8hz),134.0(d,j=10.4hz),120.0(d,j=135.9hz),114.2(d,j=13.3hz),67.1(d,j=6.8hz),55.2,44.8。
31pnmr(162mhz,cdcl3,ppm)δ=29.75.hrms(esi)calcdforc18h23no4p[m+h]+348.1365,found348.1364。
实施例9
在15ml电解池中加入双(4-氯苯基)氧化膦(0.3mmol,81.3mg)、吗啡啉(0.9mmol,78.3mg)、碘化钾(1mmol,166mg)和乙醇(10ml),恒电流30ma条件下搅拌反应。反应完成后(tlc跟踪检测),旋干得到的残留物用乙酸乙酯/甲醇体系作为洗脱剂过色谱柱得到产物3fa,产率为75%,纯度98%。
通过核磁共振波谱仪对所述加成产物进行分析,结果参见图25~27。其中,图25为本发明实施例9提供的产物1h核磁共振(1hnmr)谱图;图26为本发明实施例9提供的产物13c核磁共振(13cnmr)谱图;图27为本发明实施例9提供的产物31p核磁共振(31pnmr)谱图。由图25~27可知,本发明实施例9提供的产物结构如3fa所示。
对所述产物进行测定,其表征数据为:
1hnmr(400mhz,cdcl3,ppm):δ=7.82-7.77(m,4h),7.47-7.45(m,4h),3.71(s,4h),3.06(s,4h)。
13cnmr(100mhz,cdcl3,ppm):δ=138.9(d,j=3.3hz),133.8(d,j=9.9hz),129.2(d,j=13.0hz),129.0(d,j=131.1hz),67.1(d,j=6.5hz),45.0。
31pnmr(162mhz,cdcl3,ppm)δ=27.22.hrms(esi)calcdforc16h17cl2no2p[m+h]+356.0374,found356.0377。
实施例10
在15ml电解池中加入苯基膦酸乙酯(0.3mmol,50.7mg)、吗啡啉(0.9mmol,78.3mg)、碘化钾(1mmol,166mg)和乙醇(10ml),恒电流30ma条件下搅拌反应。反应完成后(tlc跟踪检测),旋干得到的残留物用乙酸乙酯/甲醇体系作为洗脱剂过色谱柱得到产物3ia,产率为88%,纯度99%。
通过核磁共振波谱仪对所述加成产物进行分析,结果参见图28~30。其中,图28为本发明实施例10提供的产物1h核磁共振(1hnmr)谱图;图29为本发明实施例10提供的产物13c核磁共振(13cnmr)谱图;图30为本发明实施例10提供的产物31p核磁共振(31pnmr)谱图。由图28~30可知,本发明实施例10提供的产物的结构如3ia所示。
对所述产物进行测定,其表征数据为:
1hnmr(400mhz,cdcl3,ppm):δ=7.76–7.71(m,2h),7.54–7.45(m,3h),4.22–4.09(m,2h),3.63(s,4h),3.11-3.10(m,4h),1.39(t,j=7.0hz,3h)。
13cnmr(100mhz,cdcl3,ppm):δ=131.7(d,j=2.9hz),131.1(d,j=9.4hz),129.7(d,j=172.5hz),128.3(d,j=14.0hz),66.8(d,j=5.7hz),60.5(d,j=5.9hz),40.9,16.2(d,j=6.5hz)。
31pnmr(162mhz,cdcl3,ppm)δ=21.04.hrms(esi)calcdforc12h19no3p[m+h]+256.1103,found256.1103。
由上述实施例可知,本发明在通电条件下,制备得到了取代的磷酰胺类化合物。
以上实施例的说明只是用于帮助理解本发明的方法及其核心思想。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!