一种电化学合成α‑酰氧基酮的方法与流程
【技术领域】
本发明属于有机合成领域,具体涉及一种电化学合成α-酰氧基酮的方法。
背景技术:
α-酰氧基酮化合物广泛存在于天然产物或者药物中,常常作为生理活性分子和药物分子的关键结构骨架,具有较高的生物活性和潜在的应用价值(fukuda,t.;matsumoto,a.;takahashi,y.;tomoda,h.;omura,s.j.antibiot.2005,58,252;yan,b.-f.;fang,s.-t.;li,w.-z.;liu,s.-j.;wang,j.-h.;xia,c.-h.nat.prod.res.2015,29,2013.)。随着它的α-功能化反应在有机合成过程中的作用日益凸显,其合成方法已成为近年来化学工作者研究的重点。目前,普遍报道的α-酰氧基酮化合物合成方法如下:2009年及2011年,ishihara课题组分别利用高价碘及碘联合氧化剂催化氧化的酮酸的内酯化合成α-酰氧基酮(uyanik,m.;yasui,t.;ishiharak.bioorg.med.chem.lett.2009,19,3848;uyanik,m.;suzuki,d.;yasui,t.;ishihara,k.angew.chem.int.ed.2011,50,5331.);2011年,张课题组利用ddq作为促进剂的酮酸的内酯化合成α-酰氧基酮(ding,y.;huang,z.-j.;yin,j.;lai,y.-s.;zhang,s.-b.;zhang,z.-g.;fang,l.;peng,s.-x.,zhang,y.-h.chem.commun.2011,47,9495.);2017年,huang,x报道了利用光催化剂联合氧化剂催化氧化酮酸合成α-酰氧基酮(huang,x.;liang,x.;yuan,j.;ni,z.-q.;zhou,y.-f.;pan,y.-j.org.chem.front.2017,4,163.)。
但是纵观上述已经报道的α-酰氧基酮化合物合成方法,往往效率低下,反应时间长,不利于工业操作及大规模生产,这就大大限制了其在有机合成和药物化学领域中的应用空间;同时在合成过程中通常还需要加入大量的氧化剂,这不仅存在一定的安全隐患,而且过量的氧化剂还容易造成资源浪费和环境污染。因此,研究一种操作简单、条件温和、绿色环保、稳定高效、易于控制并大规模生产的α-酰氧基酮合成方法具有十分重要的意义。
技术实现要素:
本发明要解决的问题是针对以上不足,提供一种操作简单、条件温和、绿色环保、稳定高效、易于控制并大规模生产的电化学合成α-酰氧基酮的方法。
本发明采用的技术方案如下:
一种电化学合成α-酰氧基酮的方法,包括以下步骤:
(1)向无隔膜电解槽中分别加入具有式i骨架结构的原料、催化剂、电解质和溶剂,然后插入电极,在恒电流下搅拌反应;
(2)通过薄层色谱跟踪反应,反应完成后分离提纯制得具有式ii骨架结构的α-酰氧基酮产物。
式i
式中:r1,r5,r7为c1-c12的烃基,r2,r3,r4,r6为氢或c1-c10的烃基;n为1或2。
式ii
式中,r1,r2,r3,r4,r5,r6,r7,n与式i中保持一致。
具体的,所述步骤(1)中原料通过分子内的氧化偶联合成α-酰氧基酮时,原料式i的骨架结构选自式i-1或式i-2骨架结构中的一种,所述步骤(2)中α-酰氧基酮产物对应的式ii骨架结构分别为式ii-1或式ii-2。
具体的,所述步骤(1)中原料通过分子间的氧化偶联合成α-酰氧基酮时,原料式i的骨架结构为具有式i-3骨架结构的原料酮和具有式i-4骨架结构的原料羧酸,所述步骤(2)中α-酰氧基酮产物对应的式ii骨架结构为式ii-3。
优选地,所述式i和式ii中r1,r5,r7独立地选自于c1-c8的烷基或取代的烷基、c5-c12的芳基或取代的芳基;r2,r3,r4,r6为氢或c1-c8的烷基或c4-c10的芳基或取代的芳基。
优选地,所述步骤(1)中的催化剂选自于四烷基溴化铵、四烷基碘化铵、nh4i、nai、ki、lii、mgi2、cai2中的一种或几种。
优选地,所述步骤(1)中的电解质选自于四烷基氟化铵、四烷基氯化铵、四烷基溴化铵、四烷基碘化铵、nh4i、nai、ki、lii、mgi2、cai2、nacl、nabr、kcl、kbr、四烷基四氟硼酸铵、四烷基高氯酸铵、四烷基醋酸铵中的一种或几种。
优选地,所述步骤(1)中的溶剂选自于水、甲醇、乙醇、三氟乙醇、乙腈、丙酮、乙酸乙酯、二氯甲烷、氯仿、石油醚中的一种或几种。
优选地,所述步骤(1)中电极选自于石墨电极、铂电极、银电极、玻碳电极、网状玻璃碳电极的一种或几种。
优选地,所述步骤(1)中的式i骨架结构的原料在反应液中的初始浓度为0.01-1mol/l,催化剂在反应液中的浓度为0.005-1mol/l,电解质在反应液中的浓度为0-1mol/l。
优选地,所述步骤(1)中的恒定电流密度为0.005-60ma/cm2。
优选地,所述步骤(1)中的反应温度为10-60oc。
本发明的优点:
1.本发明提供的电化学合成α-酰氧基酮的方法操作简单、条件温和、绿色环保、稳定高效、易于控制并大规模生产,原料通过电化学氧化偶联直接得到结构多样的α-酰氧基酮,更加通用直接。
2.本发明利用电化学合成α-酰氧基酮,避免了过量氧化剂的使用,减少了资源浪费和环境污染,条件更加温和,适用范围更广。
3.本发明提供的电化学合成α-酰氧基酮的方法工艺稳定性强,制备周期短,安全高效,利于规模化生产。
【附图说明】
图1为实施例1的1hnmr图;
图2为实施例1的13cnmr图。
图3为实施例2的1hnmr图;
图4为实施例2的13cnmr图。
图5为实施例3的1hnmr图;
图6为实施例3的13cnmr图。
图7为实施例4的1hnmr图;
图8为实施例4的13cnmr图。
图9为实施例5的1hnmr图;
图10为实施例5的13cnmr图。
图11为实施例6的1hnmr图;
图12为实施例6的13cnmr图。
图13为实施例7的1hnmr图;
图14为实施例7的13cnmr图。
图15为实施例8的1hnmr图;
图16为实施例8的13cnmr图。
图17为实施例9的1hnmr图;
图18为实施例9的13cnmr图。
图19为实施例10的1hnmr图;
图20为实施例10的13cnmr图。
图21为实施例11的1hnmr图;
图22为实施例11的13cnmr图。
【具体实施方式】
为了更充分理解本发明的技术内容,下面通过具体实施例对本发明技术方案进行进一步介绍和说明。以下实施例只是描述性的,不是限定性的,不能以此限定本发明的保护范围。
实施例1
向无隔膜电解槽中分别加入10g原料4-苯甲酰基丁酸,2.8g催化剂四正丁基碘化铵,12g电解质四丁基醋酸铵和50ml乙腈、2ml三氟乙醇作为混合溶剂,然后插入石墨电极,通入电流密度为10ma/cm2的恒电流在40oc下搅拌反应;通过薄层色谱跟踪反应,反应完成后在真空下旋去溶剂,然后分离得到产物5-苯甲酰基二氢呋喃-2(3h)-酮6.7g,产率68%。
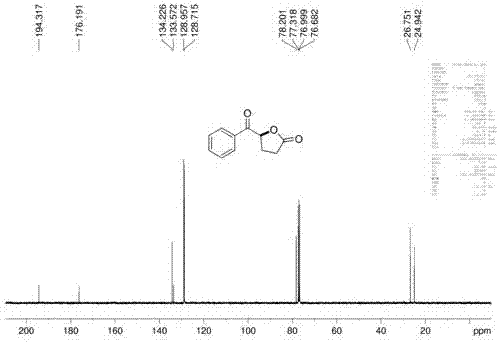
1hnmr(400mhz,cdcl3):δ7.99-7.97(d,j=7.6hz,2h),7.66-7.63(t,j=7.4hz,1h),7.54-7.50(t,j=7.6hz,2h),5.83-5.81(m,1h),2.64-2.56(m,3h),2.50-2.44(m,1h);13cnmr(100mhz,cdcl3):δ194.3,176.2,134.2,133.6,129.0,128.7,78.2,26.8,24.9。
实施例2
向无隔膜电解槽中分别加入234mg原料4-(2,4,6-三甲基苯甲酰基)丁酸,738mg催化剂四正丁基碘化铵和8ml乙腈、2ml甲醇作为混合溶剂,然后插入铂片电极,通入电流密度为8ma/cm2的恒电流在40oc下搅拌反应;通过薄层色谱跟踪反应,反应完成后在真空下旋去溶剂,然后分离得到产物5-(2,4,6-三甲基苯甲酰基)二氢呋喃-2(3h)-酮174mg,产率75%。
1hnmr(400mhz,cdcl3):δ6.88(s,2h),5.28-5.24(dd,j=5.0hz,8.3hz,1h),2.61-2.45(m,3h),2.37-2.33(m,1h),2.29(s,3h),2.24(s,6h);13cnmr(100mhz,cdcl3):δ205.7,176.1,140.0,135.0,134.1,128.8,81.7,26.9,24.6,21.0,19.5。
实施例3
向无隔膜电解槽中分别加入198mg原料5-氧-5-(2-噻吩基)戊酸,750mg催化剂nai,5ml溶剂乙醇,然后插入石墨电极,通入电流密度为6ma/cm2的恒电流在60oc下搅拌反应;通过薄层色谱跟踪反应,反应完成后在真空下旋去溶剂,然后分离得到产物5-(噻吩-2-羰基)二氢呋喃-2(3h)-酮153mg,产率78%。
1hnmr(400mhz,cdcl3):δ7.92-7.91(d,j=3.2hz,1h),7.79-7.77(d,j=4.7hz,1h),7.22-7.19(t,j=4.3hz,1h),5.59-5.56(m,1h),2.67-2.55(m,3h),2.53-2.48(m,1h);13cnmr(100mhz,cdcl3):δ187.9,176.1,140.1,135.6,133.9,128.6,79.2,26.8,25.4。
实施例4
向无隔膜电解槽中分别加入297mg原料(e)-7-(4-溴苯基)-5-氧-6-烯庚酸,37mg催化剂四正丁基碘化铵,684mg电解质四丁基高氯酸铵,20ml溶剂乙睛,然后插入石墨电极,通入电流密度为20ma/cm2的恒电流在30oc下搅拌反应;通过薄层色谱跟踪反应,反应完成后在真空下旋去溶剂,然后分离得到产物(e)-5-(3-(4-溴苯基)丙烯酰基)二氢呋喃-2(3h)-酮207mg,产率70%。
1hnmr(400mhz,cdcl3):δ7.73-7.69(d,j=16.0hz,1h),7.56-7.54(d,j=8.4hz,2h),7.48-7.46(d,j=8.4hz,2h),7.07-7.03(d,j=16.0hz,1h),5.11-5.08(t,j=6.8hz,1h),2.62-2.55(m,3h),2.43-2.35(m,1h);13cnmr(100mhz,cdcl3):δ195.3,176.1,144.7,132.8,132.3,130.1,125.7,120.4,81.2,27.3,24.9。
实施例5
向无隔膜电解槽中分别加入241mg原料6-(2-氯苯基)-5-氧己酸,15mg催化剂nh4i,580mg电解质nacl,10ml溶剂乙睛,然后插入石墨电极,通入电流密度为30ma/cm2的恒电流在10oc下搅拌反应;通过薄层色谱跟踪反应,反应完成后在真空下旋去溶剂,然后分离得到产物5-(2-(2-氯苯基)乙酰基)二氢呋喃-2(3h)-酮134mg,产率56%。
1hnmr(400mhz,cdcl3):δ7.67-7.65(d,j=7.9hz,1h),7.48-7.44(t,j=7.5hz,1h),7.33-7.31(m,2h),5.71-5.68(m,1h),2.62-2.57(m,3h),2.53-2.51(m,3h),2.41-2.38(m,1h);13cnmr(100mhz,cdcl3):δ197.8,176.2,139.9,133.7,132.6,132.4,129.0,125.9,79.3,26.9,25.1,21.4。
实施例6
向无隔膜电解槽中分别加入110mg原料3,3-二甲基-5-氧-5-(对甲苯基)戊酸,48mg催化剂四正丁基溴化铵,3g电解质四正丁基四氟硼酸铵,50ml溶剂水,然后插入石墨电极,通入电流密度为60ma/cm2的恒电流在40oc下搅拌反应;通过薄层色谱跟踪反应,反应完成后在真空下旋去溶剂,然后分离得到产物4,4-二甲基-5-(4-甲基苯基)二氢呋喃-2(3h)-酮49mg,产率42%。
1hnmr(400mhz,cdcl3):δ7.85-7.83(d,j=8.0hz,2h),7.33-7.31(d,j=8.0hz,2h),5.51(s,1h),2.61-2.56(d,j=17.0hz,1h),2.44(s,3h),2.34-2.30(d,j=17.1hz,1h),1.38(s,3h),0.97(s,3h);13cnmr(100mhz,cdcl3):δ195.1,175.8,145.4,133.3,129.7,128.6,85.3,41.8,40.5,28.3,23.5,21.7。
实施例7
向无隔膜电解槽中分别加入220mg原料6-氧-6-(对甲苯基)己酸,30mg催化剂四甲基碘化铵,775mg电解质四正丁基六氟磷酸铵,20ml溶剂乙腈,然后插入网状玻璃碳电极,通入电流密度为0.005ma/cm2的恒电流在40oc下搅拌反应;通过薄层色谱跟踪反应,反应完成后在真空下旋去溶剂,然后分离得到产物6-(4-甲基苯甲酰基)四氢-2h-吡喃-2-酮87mg,产率40%。
1hnmr(400mhz,cdcl3):δ7.86-7.84(d,j=8.0hz,2h),7.31-7.29(d,j=8.0hz,2h),5.89-5.86(t,j=5.2hz,1h),2.69-2.56(m,2h),2.43(s,3h),2.26-2.19(m,1h),2.10-2.05(m,1h),1.88-1.80(m,2h);13cnmr(100mhz,cdcl3):δ194.7,170.0,145.3,131.0,129.7,128.7,79.2,29.4,24.6,21.7,17.0。
实施例8
向无隔膜电解槽中分别加入218mg原料3-(1-氧-1,2,3,4-四氢化萘-2-基)丙酸,30mg催化剂四甲基碘化铵,600mg电解质四正丁基醋酸铵,18ml溶剂乙腈、1ml三氟乙醇,然后插入石墨电极,通入电流密度为8ma/cm2的恒电流在40oc下搅拌反应;通过薄层色谱跟踪反应,反应完成后在真空下旋去溶剂,然后分离得到产物3,3',4,4'-四氢-1'h,5h-螺[呋喃-2,2'-萘]-1',5-二酮214mg,产率99%。
1hnmr(400mhz,cdcl3):δ8.08-8.06(d,j=7.8hz,1h),7.57-7.53(t,j=7.4hz,1h),7.39-7.35(t,j=7.5hz,1h),7.29-7.27(d,j=8.1hz,1h),3.23-3.17(m,1h),3.13-3.05(m,1h),2.84-2.74(m,1h),2.65-2.53(m,3h),2.34-2.28(m,1h),2.20-2.12(dd,j=10.7hz,23.0hz,1h);13cnmr(100mhz,cdcl3):δ193.5,176.1,143.0,134.4,129.9,128.7,128.5,127.3,85.0,34.4,29.6,27.8,25.7。
实施例9
向无隔膜电解槽中分别加入218mg原料3-(5-甲基-1-氧-2,3-二氢-1h-茚-2-基)丙酸,37mg催化剂四正丁基碘化铵,775mg电解质四正丁基六氟磷酸铵,20ml溶剂乙腈,然后插入铂片电极,通入电流密度为20ma/cm2的恒电流在30oc下搅拌反应;通过薄层色谱跟踪反应,反应完成后在真空下旋去溶剂,然后分离得到产物5'-甲基-3,4-二氢-5h-螺[呋喃-2,2'-茚]-1',5(3'h)-二酮199mg,产率92%。
1hnmr(400mhz,cdcl3):δ7.58(s,1h),7.51-7.49(d,j=7.8hz,1h),7.36-7.34(d,j=7.8hz,1h),3.56-3.52(d,j=17.2hz,1h),3.32-3.27(d,j=17.2hz,1h),3.03-2.98(m,1h),2.70-2.62(m,1h),2.52-2.45(m,1h),2.42(s,3h),2.31-2.23(m,1h);13cnmr(100mhz,cdcl3):δ201.5,176.2,147.5,138.5,137.7,133.1,126.2,124.9,86.7,39.3,31.2,28.5,21.0。
实施例10
向无隔膜电解槽中分别加入原料120mg苯乙酮,240mg乙酸,37mg催化剂四正丁基碘化铵,600mg电解质四正丁基醋酸铵,20ml溶剂乙腈,然后插入银电极,通入电流密度为10ma/cm2的恒电流在40oc下搅拌反应;通过薄层色谱跟踪反应,反应完成后在真空下旋去溶剂,然后分离得到产物2-氧-2-苯乙基乙酸71mg,产率40%。
1hnmr(400mhz,cdcl3):δ7.93-7.91(d,j=7.2hz,2h),7.63-7.7.60(t,j=6.4hz,1h),7.51-7.47(t,j=7.6hz,2h),5.35(s,2h),2.24(s,3h);13cnmr(100mhz,cdcl3):δ192.1,170.4,134.2,133.9,128.8,127.7,66.0,20.6。
实施例11
向无隔膜电解槽中分别加入原料134mg苯丙酮,366mg苯甲酸,37mg催化剂四正丁基碘化铵,600mg电解质四正丁基醋酸铵,20ml溶剂乙腈,然后插入铂片电极,通入电流密度为20ma/cm2的恒电流在40oc下搅拌反应;通过薄层色谱跟踪反应,反应完成后在真空下旋去溶剂,然后分离得到产物1-氧-1-苯丙-2-基苯甲酸酯89mg,产率35%。
1hnmr(400mhz,cdcl3):δ8.1-8.08(d,j=7.6hz,2h),8.02-8.00(d,j=7.6hz,2h),7.61-7.56(m,2h),7.51-7.43(m,4h),6.24-6.18(q,j=7.0hz,1h),1.68-1.67(d,j=7.0hz,3h);13cnmr(100mhz,cdcl3):δ196.7,166.0,134.5,133.6,133.3,129.9,129.5,128.8,128.5,128.4,71.9,17.2。
以上所述实施例仅表达了本发明的实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
- 还没有人留言评论。精彩留言会获得点赞!