一种盐酸右美托咪定粗品中异构体的检测方法与流程
本发明属于药物检测技术领域,具体来说,涉及到一种盐酸右美托咪定粗品中异构体的检测方法。
背景技术:
盐酸右美托咪定是一种全新的α2-肾上腺素受体激动剂,具有强效镇静作用,是可乐定作用的8倍。主要作用于脑桥和延髓以及脑干篮斑的肾上腺素受体,具有抗交感疗效,半衰期2.3h,镇静效果强大兼具其他麻醉辅助作用。主要适用于重症监护治疗期间最初插管和使用呼吸机病人的镇静,可在机械通气患者拔管之前、拔管中和拔管后连续使用,在拔管前无需停药;也可用于非插管患者在尹术和其他操作前和/或术中的镇静。在美国应用5年多的临床经验表明,盐酸右美托咪定可产生稳定的镇定和觉醒作用,对重症病人的生理及心理方面的需求有独特的协同作用,可明显减少诱导麻醉所需的麻醉剂用量;术前给予本品可减少术前和术后的阿片或非阿片类止痛剂的用量,这一特性对于麻醉和重症监护有重要的意义;还可以促进儿茶酚胺血流动力学的稳定性,有效减轻气管插管、手术应激和麻醉及恢复早期血流动力学应答。
盐酸右美托咪定的对映体左旋异构体在抗交感、镇痛、镇静方面几乎没有作用,因此控制盐酸右美托咪定中左旋异构体含量非常重要。高存强建立了HPLC法测定盐酸右美托咪定及注射剂中的R-异构体:采用手性色谱柱Chiral-AGP(4.0mm×150mm,5μm)色谱柱;流动相:0.03mol/L磷酸氢二钾缓冲液-乙腈(82.5:17.5);检测波长:214nm;流速:0.5ml/min。该法结果:盐酸右美托咪定与R-异构体分离良好,最低检测量为0.4ng,R-异构体的回收率为98.0%。但是,上述测定方法专属性差,灵敏度不够高,不适宜在盐酸右美托咪定粗品中对异构体进行检测。
技术实现要素:
为解决上述技术问题,本发明提供了一种专属性强、灵敏度高的盐酸右美托咪定粗品中异构体的检测方法。
本发明所述的一种盐酸右美托咪定粗品中异构体的检测方法,所述测定方法具体步骤为:采用高效液相色谱法进行进检测,色谱柱:手性柱色谱柱或N-氧化石松碱M键合硅胶柱;流速:0.2-2.0ml/min;检测波长:180-380nm;柱温:0-40℃;流动相:正己烷-乙醇-有机酸和/或有机碱。
本发明所述的一种盐酸右美托咪定粗品中异构体的检测方法,所述色谱柱选择AS-H型手性柱或CHIRALPAK手性柱;所述流速为0.5-1.5ml/min;所述柱温为20-30℃;所述流动相中有机酸和/或有机碱为甲酸、乙酸、丙酸、冰醋酸、三氟乙酸、乙二胺、二乙胺、三乙胺中的一种或多种。
本发明所述的一种盐酸右美托咪定粗品中异构体的检测方法,所述检测波长为200-300nm;所述流动相中有机酸和/或有机碱为甲酸、乙酸、三氟乙酸、二乙胺、三乙胺中的一种或多种。
本发明所述的一种盐酸右美托咪定粗品中异构体的检测方法,所述流动相比例为正己烷-乙醇-有机酸-有机碱(85-99):(15-1):(0-1):(0-1)。
本发明所述的一种盐酸右美托咪定粗品中异构体的检测方法,所述流动相比例为正己烷-乙醇-有机酸-有机碱(90-95):(10-5):(0-1):(0-1)。
本发明所述的一种盐酸右美托咪定粗品中异构体的检测方法,所述色谱柱为AS-H型手性柱;所述检测波长为210nm;所述柱温为25℃;所述流动相比例为正己烷-乙醇-三氟乙酸-二乙胺(95:5:0.3:0.05)。
本发明所述的一种盐酸右美托咪定粗品中异构体的检测方法,所述色谱柱为N-氧化石松碱M键合硅胶柱;所述检测波长为210nm;所述柱温为25℃;所述流动相比例为正己烷-乙醇-三氟乙酸-二乙胺(95:5:0.3:0.05)。
本发明所述的一种盐酸右美托咪定粗品中异构体的检测方法,所述N-氧化石松碱M键合硅胶柱的制备步骤为:1)取20g活化的硅胶于500mL三口瓶中,再量取100mL甲苯和40mL的γ-氨丙基三乙氧基硅烷,120℃下油浴,搅拌回流13h,待冷却后,产物依次用甲苯,乙醇洗涤抽滤至无胶状物存在;在60℃下干燥24h,干燥箱内冷却;2)在500mL的单口瓶中加入步骤1)的产物,然后依次加入120mL 35%的甲醛溶液和2mL的乙酸溶液,室温搅拌反应2h,经无水乙醇抽滤后,置于500mL三口瓶中备用;取N-氧化石松碱M 4g溶于适量水溶液中,调节溶液的pH值至8.0,加入上述三口瓶中,60℃搅拌下反应10h;待产物冷却,依次用大量二次水、无水乙醇洗涤抽滤至滤液清亮;60℃恒温干燥6h,于干燥箱内冷却后装棕色瓶内得N-氧化石松碱M键合硅胶;3)将N-氧化石松碱M键合硅胶常规装柱,即得。
测定过程:取美托咪定对照品20mg,置50ml量瓶中,加乙醇超声处理使溶解并稀释至刻度,摇匀,精密量取2ml置10ml量瓶中,加乙醇稀释至刻度,摇匀,作为预试液。取预试液20μl注入液相色谱仪,记录色谱图,主峰依次为右美托咪定与左美托咪定,右美托咪定与左美托咪定的分离度应符合要求。取本品20mg,置50ml量瓶中,加乙醇溶解并稀释至刻度,摇匀,作为供试品溶液;精密量取供试品溶液1ml置100ml量瓶中,加乙醇稀释至刻度,摇匀,作为对照溶液。精密量取对照溶液20μl注入液相色谱仪,调节检测灵敏度,使主成分的峰高达满量程的10%~20%;精密量取供试品溶液和对照溶液各20μl注入液相色谱仪,记录色谱图;供试品溶液中如显左美托咪定峰,左美托咪定峰的峰面积不得大于对照溶液主峰面积(1.0%)。
与现有技术相比,本发明所述盐酸右美托咪定粗品中异构体的检测方法专属性好、准确度高,主峰与异构体能够有效的分离,能够准确地检测出盐酸右美托咪定粗品中的异构体,为该产品的质量控制及建立统一的标准建立了良好的推进作用。
附图说明
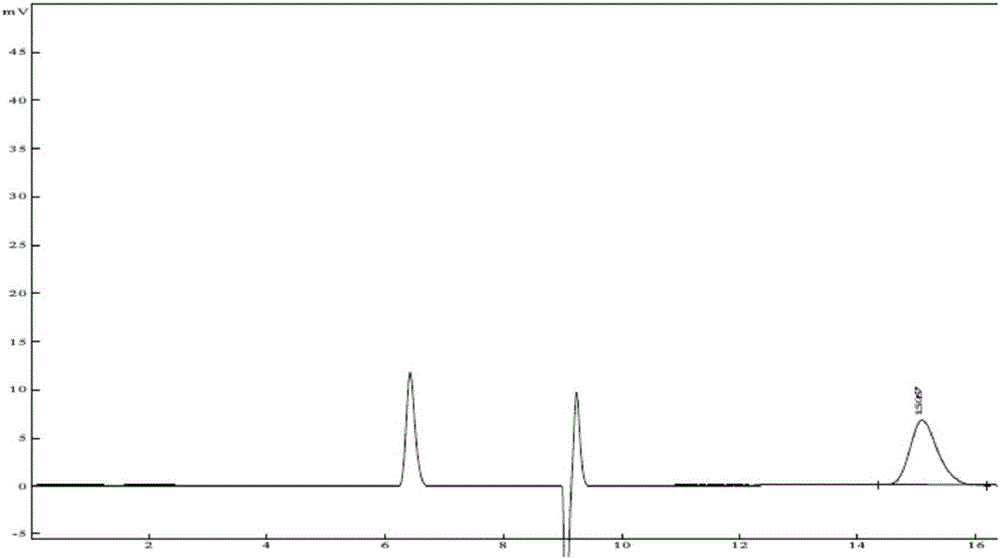
图1是盐酸右美托咪定专属性供试品溶液图谱;图2是盐酸右美托咪定专属性对照品溶液图谱;图3是盐酸右美托咪定专属性系统适用性溶液图谱;图4是盐酸右美托咪定检测限图谱;图5是盐酸右美托咪定141203101批供试品溶液图谱;图6是盐酸右美托咪定141203102批供试品溶液图谱;图7是盐酸右美托咪定141203103批供试品溶液图谱。
具体实施方式
下面结合具体的实施例对本发明所述盐酸右美托咪定粗品中异构体的检测方法做进一步说明,但是本发明的保护范围并不限于此。
实施例1
盐酸右美托咪定粗品中异构体的测定是采用高效液相色谱法,其色谱条件为色谱柱:N-氧化石松碱M键合硅胶柱;流速:0.5ml/min检测波长:210;柱温:25℃;流动相:正己烷-乙醇-三氟乙酸-二乙胺(95:5:0.3:0.05)。
所述N-氧化石松碱M键合硅胶柱的制备步骤为:1)取20g活化的硅胶于500mL三口瓶中,再量取100mL甲苯和40mL的γ-氨丙基三乙氧基硅烷,120℃下油浴,搅拌回流13h,待冷却后,产物依次用甲苯,乙醇洗涤抽滤至无胶状物存在;在60℃下干燥24h,干燥箱内冷却;2)在500mL的单口瓶中加入步骤1)的产物,然后依次加入120mL 35wt%的甲醛溶液和2mL的乙酸,室温搅拌反应2h,经无水乙醇抽滤后,置于500mL三口瓶中备用;取N-氧化石松碱M 4g溶于适量水溶液中,调节溶液的pH值至8.0,加入上述三口瓶中,60℃搅拌下反应10h;待产物冷却,依次用大量二次水、无水乙醇洗涤抽滤至滤液清亮;60℃恒温干燥6h,于干燥箱内冷却后装棕色瓶内得N-氧化石松碱M键合硅胶;3)将N-氧化石松碱M键合硅胶常规装柱,即得。
稀释剂:乙醇(色谱级)。系统适用性试验:取美托咪定对照品20mg,置50ml量瓶中,加乙醇超声处理使溶解并稀释至刻度,摇匀,精密量取2ml置10ml量瓶中,加乙醇稀释至刻度,摇匀,作为预试液。取预试液20μl注入液相色谱仪,记录色谱图,主峰依次为右美托咪定与左美托咪定,右美托咪定与左美托咪定的分离度应符合要求。
测定方法:取本品20mg,置50ml量瓶中,加乙醇溶解并稀释至刻度,摇匀,作为供试品溶液;精密量取供试品溶液1ml置100ml量瓶中,加乙醇稀释至刻度,摇匀,作为对照溶液。精密量取对照溶液20μl注入液相色谱仪,调节检测灵敏度,使主成分的峰高达满量程的10%~20%;精密量取供试品溶液和对照溶液各20μl注入液相色谱仪,记录色谱图;供试品溶液中如显左美托咪定峰,左美托咪定峰的峰面积不得大于对照溶液主峰面积(1.0%)。
本发明中所述流动相的配比为常温下的体积比。为证实该方法准确可靠,精密度高,能够用于控制盐酸右美托咪定粗品中的异构体,进行了如下方法验证。
1.1专属性
系统适用性试验样品中右美托咪定与异构体之间完全分离,分离度为3.65,理论塔板数为4582,分离效果和柱效均良好。见图1-3。
1.2检测限
取供试品溶液,采用逐级稀释法,依次进样。当信噪比(S/N)约为3时,此对应的盐酸右美托咪定为检测限为0.2ng。见图4。
1.3线性
取盐酸右美托咪定20mg,置50ml量瓶中,加乙醇溶解并稀释至刻度,摇匀,作为贮备液;分别精密量取1、1.5、2、2.5、3ml置于200ml量瓶中,分别加乙醇溶解并稀释至刻度,摇匀。分别取上述5份溶液20μl注入液相色谱仪,结果见下表。
1.4样品测定
取盐酸右美托咪定粗品依法测定三批样品的异构体,记录色谱图。结果采用本发明检测方法,检出限为0.2ng,线性较好R为0.9999,异构体检测结果见下表及见图5-7。
实施例2
盐酸右美托咪定粗品中异构体的测定是采用高效液相色谱法,其色谱条件为色谱柱:AS-H型手性柱;流速:0.5ml/min检测波长:210;柱温:25℃;流动相:正己烷-乙醇-三氟乙酸-二乙胺(95:5:0.3:0.05)。
稀释剂:乙醇(色谱级)。系统适用性试验:取美托咪定对照品20mg,置50ml量瓶中,加乙醇超声处理使溶解并稀释至刻度,摇匀,精密量取2ml置10ml量瓶中,加乙醇稀释至刻度,摇匀,作为预试液。取预试液20μl注入液相色谱仪,记录色谱图,主峰依次为右美托咪定与左美托咪定,右美托咪定与左美托咪定的分离度应符合要求。
测定方法:取本品20mg,置50ml量瓶中,加乙醇溶解并稀释至刻度,摇匀,作为供试品溶液;精密量取供试品溶液1ml置100ml量瓶中,加乙醇稀释至刻度,摇匀,作为对照溶液。精密量取对照溶液20μl注入液相色谱仪,调节检测灵敏度,使主成分的峰高达满量程的10%~20%;精密量取供试品溶液和对照溶液各20μl注入液相色谱仪,记录色谱图;供试品溶液中如显左美托咪定峰,左美托咪定峰的峰面积不得大于对照溶液主峰面积(1.0%)。
通过方法学验证,采用AS-H型手性柱时,同洗脱条件下,分离柱效与N-氧化石松碱M键合硅胶柱差异不大,但其容易受到洗脱条件的影响。
- 还没有人留言评论。精彩留言会获得点赞!