肿瘤‑标志物的组的制作方法
本发明涉及癌症诊断以及为此的诊断方式领域。
甲状腺结节为碘缺乏地区的地方病,如欧洲的高山地区,在那里其流行率为10-20%。其根据其组织学,分类为2种良性类型——结节性甲状腺肿(Struma nodosa,SN)和滤泡性甲状腺腺瘤(Follicular Thyroid Adenoma,FTA),以及恶性实体——滤泡性甲状腺癌(Follicular Thyroid Carcinoma,FTC),乳突状甲状腺癌(Papillary Thyroid Carcinoma,PTC),髓样甲状腺癌(Medullary Thyroid Carcinoma,MTC)及未分化甲状腺癌(Anaplastic Thyroid Carcinoma,ATC)。传统地,良性和恶性甲状腺结节之间的区分是通过闪烁扫描法以及细针抽吸后进行组织学检验而完成。尽管在甲状腺结节和甲状腺癌的诊断和治疗上有很多进展,这些方法缺少特异性是众人皆知的,尤其在区分FTA和FTC上,这导致大量的病人不必要地被当作恶性疾病治疗。
由于先前的方法有诊断限制性,尤其是细针抽吸后进行细胞学检验,许多研究者已经进行了表达谱研究,希望鉴定出新的诊断工具。这些分析尝试用大规模转录水平表达谱技术如cDNA微阵列、寡核苷酸阵列以及基因表达系列分析(Serial Analysis of Gene Expression,SAGE)鉴定在疾病发展或进程中具有重要作用的差异性表达蛋白质。典型地,鉴定出数十或数百种基因,其中许多预期为假阳性,只有一小部分能用作诊断/预后标志物或治疗靶标(Griffith等人,J Clin Oncol 24(31):5043-5051(2006))。
在其他类型的癌症中,已经显示基因表达谱能为区分不同临床相关的肿瘤实体增添重要价值。例如US 2006/183141 A描述了来自核心血清应答签名(core serum response signature)的肿瘤标志物的分类。不同研究已经尝试基于基因表达谱为不同甲状腺癌实体分类,每项研究都对5种实体中的2种进行区分。然而,这些研究没有或很少有共同的基因,而且将来自一项研究的分类物应用于来自另一项研究的数据一般产生很差的分类结果。
本发明的一个目标是提供可靠的区别性标志物用于癌症的诊断,尤其是辨别良性甲状腺结节和恶性滤泡性甲状腺癌(FTC)以及乳突状甲状腺癌(PTC)。
因此,本发明提供了特异于至少3种肿瘤标志物的部分的组,所述肿瘤标志物选自肿瘤标志物PI-1到PI-33,PII-1到PII-64,PIII-1到PIII-70,fi-1到fi-147,PIV-1到PIV-9,优选地为PIV-4或PIV-5,以及PV-1到PV-11,优选地为PV-1,PV-2和PV-4到PV-11。这些肿瘤标志物涉及肿瘤中异常表达的不同基因,并且在表1-6中给出,能通过其基因鉴定标记、其描述性基因名称而对其鉴定,但最清楚的是通过其UniGeneID或其在常用序列数据库如NCBI GenBank,EMBL-EBI数据库,EnsEMBL或日本DNA数据库中参考特定序列的登记号。这些标志物已经以优选组(PI到PV,FI)的形式鉴定出来,但能以任何形式作为发明组的靶标而合并。
表1:PTC标志物组PI-1到PI-33
表2:PTC标志物组PII-1到PII-64
表3:PTC标志物组PIII-1到PIII-70
表4:FTC标志物组FI-1到FI-147
表5:PTC标志物组PIV-1到PIV-9
表6:PTC标志物组PV-1到PV-11
本发明的组能用于检测癌症或肿瘤细胞,尤其是甲状腺癌,甚至可用于区分良性甲状腺结节和恶性滤泡性甲状腺癌(FTC)以及乳突状甲状腺癌(PTC)。在优选的实施方式中,组包括特异于至少3种肿瘤标志物的部分,所述肿瘤标志物选自肿瘤标志物PI-1到PI-33,PII-1到PII-64,PIII-1到PIII-70,以及PIV-1到PIV-9,优选地为PIV-4或PIV-5以及PV-1到PV-11,优选地为PV-1、PV-2以及PV-4到PV-11,尤其选自肿瘤标志物PI-1到PI-33。这些标志物对乳突状甲状腺瘤(PTC)和经诊断被定性为PTC的甲状腺癌具有特异性。
在类似的优选实施方式中,组包括特异于选自肿瘤标志物FI-1到FI-147的至少3种肿瘤标志物的部分。这些标志物对滤泡性甲状腺瘤(FTC)和经诊断被定性为FTC的甲状腺癌具有特异性。
尤其优选地,组包括特异于肿瘤标志物SERPINA1(丝氨酸(或半胱氨酸)蛋白酶抑制子,A分枝(α-1抗蛋白酶,抗胰蛋白酶),成员1;NM_000295,NM_001002236,NM_001002235)的部分,其为PTC的一种非常有效的标志物。此标志物作为该组的单个成员能区分PTC和良性状态。
优选地,组包括至少5种或至少10种,优选地至少15种,更优选地至少20中,尤其优选地至少25种,最优选地至少30种特异于以上表1-6的肿瘤标志物的部分。组可选自特异于任何至少3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,25,30,33,35,40,45,50,55,60,64,65,70,75,80,85,90,95,100,110,120,130,140,145,147,150,160,170,180,190或200种以上肿瘤标志物的部分,例如,选自PI-1到PI-33,PII-1到PII-64,PIII-1到PIII-70,FI-1到FI-147,PIV-1到PIV-9,优选地PIV-4或PIV-5,及PV-1到PV-11,优选地PV-1,PV-2及PV-4到PV-11,具体而言选自PI-1,PI-2,PI-3,PI-4,PI-5,PI-6,PI-7,PI-8,PI-9,PI-10,PI-11,PI-12,PI-13,PI-14,PI-15,PI-16,PI-17,PI-18,PI-19,PI-20,PI-21,PI-22,PI-23,PI-24,PI-25,PI-26,PI-27,PI-28,PI-29,PI-30,PI-31,PI-32,PI-33,PII-1,PII-2,PII-3,PII-4,PII-5,PII-6,PII-7,PII-8,PII-9,PII-10,PII-11,PII-12,PII-13,PII-14,PII-15,PII-16,PII-17,PII-18,PII-19,PII-20,PII-21,PII-22,PII-23,PII-24,PII-25,PII-26,PII-27,PII-28,PII-29,PII-30,PII-31,PII-32,PII-33,PII-34,PII-35,PII-36,PII-37,PII-38,PII-39,PII-40,PII-41,PII-42,PII-43,PII-44,PII-45,PII-46,PII-47,PII-48,PII-49,PII-50,PII-51,PII-52,PII-53,PII-54,PII-55,PII-56,PII-57,PII-58,PII-59,PII-60,PII-61,PII-62,PII-63,PII-64,PIII-1,PIII-2,PIII-3,PIII-4,PIII-5,PIII-6,PIII-7,PIII-8,PIII-9,PIII-10,PIII-11,PIII-12,PIII-13,PIII-14,PIII-15,PIII-16,PIII-17,PIII-18,PIII-19,PIII-20,PIII-21,PIII-22,PIII-23,PIII-24,PIII-25,PIII-26,PIII-27,PIII-28,PIII-29,PIII-30,PIII-31,PIII-32,PIII-33,PIII-34,PIII-35,PIII-36,PIII-37,PIII-38,PIII-39,PIII-40,PIII-41,PIII-42,PIII-43,PIII-44,PIII-45,PIII-46,PIII-47,PIII-48,PIII-49,PIII-50,PIII-51,PIII-52,PIII-53,PIII-54,PIII-55,PIII-56,PIII-57,PIII-58,PIII-59,PIII-60,PIII-61,PIII-62,PIII-63,PIII-64,PIII-65,PIII-66,PIII-67,PIII-68,PIII-69,PIII-70,FI-1,FI-2,FI-3,FI-4,FI-5,FI-6,FI-7,FI-8,FI-9,FI-10,FI-11,FI-12,FI-13,FI-14,FI-15,FI-16,FI-17,FI-18,FI-19,FI-20,FI-21,FI-22,FI-23,FI-24,FI-25,FI-26,FI-27,FI-28,FI-29,FI-30,FI-31,FI-32,FI-33,FI-34,FI-35,FI-36,FI-37,FI-38,FI-39,FI-40,FI-41,FI-42,FI-43,FI-44,FI-45,FI-46,FI-47,FI-48,FI-49,FI-50,FI-51,FI-52,FI-53,FI-54,FI-55,FI-56,FI-57,FI-58,FI-59,FI-60,FI-61,FI-62,FI-63,FI-64,FI-65,FI-66,FI-67,FI-68,FI-69,FI-70,FI-71,FI-72,FI-73,FI-74,FI-75,FI-76,FI-77,FI-78,FI-79,FI-80,FI-81,FI-82,FI-83,FI-84,FI-85,FI-86,FI-87,FI-88,FI-89,FI-90,FI-91,FI-92,FI-93,FI-94,FI-95,FI-96,FI-97,FI-98,FI-99,FI-100,FI-101,FI-102,FI-103,FI-104,FI-105,FI-106,FI-107,FI-108,FI-109,FI-110,FI-111,112,FI-113,FI-114,FI-115,FI-116,FI-117,FI-118,FI-119,FI-120,FI-121,FI-122,FI-123,FI-124,FI-125,FI-126,FI-127,FI-128,FI-129,FI-130,FI-131,FI-132,FI-133,FI-134,FI-135,FI-136,FI-137,FI-138,FI-139,FI-140,FI-141,FI-142,FI-143,FI-144,FI-145,FI-146,FI-147,PIV-1,PIV-2,PIV-3,PIV-4,PIV-5,PIV-6,PIV-7,PIV-8,PIV-9,PV-1,PV-2,PV-3,PV-4,PV-5,PV-6,PV-7,PV-8,PV-9,PV-10,PV-11的任何一个。优选地,组特异于选自PI,PII,PIII,PIV,PV或FI的任何完整亚组。然而,还可能从这些亚组或合并的组中挑出任何小的数量,因为还能以可接受的确定性进行良性和恶性状态之间的区别或癌症的诊断。例如在一个优选的实施方式中,发明的组包括至少5种(或以上提到的任何数量)的特异于选自FI-1到FI-147的肿瘤标志物的部分。图4和5显示对于PTC和FTC的这种诊断分类概率。例如,特异于来自表2(PII亚组)任何数量的组,特异于5种标志物,只有4%的误差容限,即所有情况中96%会正确分类。用至少20个成员达到1%的误差值(99%确定性)。在FTC特异性标志物的情况下,用选自FI亚组的至少11种不同的标志物能达到稳定的8%的误差值。
根据本发明的部分为适合于特异性识别发明的标志物的分子。这种分子识别可以是在核苷酸、肽或蛋白质水平上的。优选地,所述部分为特异于肿瘤标志物核酸的核酸,尤其是寡核苷酸或引物。在另一种实施方式中,所述部分为抗体(单克隆或多克隆)或抗体片段,优选地选自Fab,Fab'Fab2,F(ab')2或scFv(单链可变片段),其特异于肿瘤标志物蛋白质。根据本发明,只要促进分子识别,核酸的哪个序列部分或蛋白质的哪些表位被该部分识别并不是至关重要的。本领域已知的部分,尤其是本文引用的参考文献(其全部作为参考并入本文)中公开的,都是合适的。
在一个优选的实施方式中,将组的部分固定在固相支持物上,优选地以微阵列或纳米阵列的形式。术语“微阵列”,同样地“纳米阵列”,用于描述微观排列的阵列(纳米阵列用于纳米规模的阵列)或指包括这种阵列的载体。两个定义不互相矛盾,且能在本发明的意义中应用。优选地,组在芯片上提供,在其上对部分进行固定。芯片可以是合适于生物分子例如部分的固定化的任何材料,包括玻璃修饰的玻璃(乙醛化修饰)或金属芯片。
根据本发明,提供了特异地用于肿瘤诊断的组。然而还可能提供更大的组,其中包括用于其他目的附加部分,具体而言在微阵列建立中,其中可能固定化大量寡核苷酸。然而优选的是提供有成本效益的组,其中包括用于单一目的的有限数量的部分。
因此,在一个优选的实施方式中,组包括至少10%,至少15%,至少20%,至少25%,至少30%,至少35%,至少40%,至少45%,至少50%,至少55%,至少60%,至少65%,至少70%,至少75%,至少80%,至少85%,至少90%,至少95%,尤其优选地至少100%的组的总分析物结合部分,其为特异于选自PI-1到PI-33,PII-1到PII-64,PIII-1到PIII-70,FI-1到FI-147,PIV-1到PIV-9,以及PV-1到PV-11(所有标志物在以上表1-6中公开),或选自PI-1到PI-33,PII-1到PII-64,PIII-1到PIII-70,FI-1到FI-147,PIV-1到PIV-9,PV-1到PV-11的任何一个的组中至少一组或其任何组合的肿瘤标志物的部分。这些优选的组合为例如组PI-1到PI-33,PII-1到PII-64,PIII-1到PIII-70,PIV-1到PIV-9,以及PV-1到PV-11中的所有标志物,尤其适合于PTC诊断。如本文使用的,“分析物结合部分”指能用于特异性检测标志物的所有部分,所述标志物具体而言是标志物基因或基因产物,包括mRNA或表达的蛋白质。基因优选地为哺乳类的基因,具体而言为人的基因。部分包括在能有多种诊断靶标的任何“分析物结合部分”的该类术语中。例如,在微阵列的实施方式中,阵列包括至少10%的特异于本发明标志物的寡核苷酸。根据目前的技术,用于在芯片上的基因(核酸分子,如分别为DNA-EST或互补DNA-EST)的检测方式使得阵列设计更简单,更强大,使用DNA分子(用于检测样品中表达的mRNA)的基因芯片为本发明的优选的实施方式。这些基因芯片还使得能够检测大量的基因产品,而使用蛋白质芯片检测(例如抗体芯片)大量的蛋白质更困难。蛋白质检测通常使用ELISA技术(即,基于-微量滴定板、珠子或芯片的ELISA)作为蛋白质芯片的一种实施方式进行。蛋白质芯片可包括用于特异性结合来自根据表1-6的列表的基因的基因产品的合适方式,例如亲和分子如单克隆或多克隆抗体或凝集素。
在进一步的实施方式中,组包括多至50000种分析物结合部分,优选地多至40000,多至35000,多至30000,多至25000,多至20000,多至15000,多至10000,多至7500,多至5000,多至3000,多至2000,多至1000,多至750,多至500,多至400,多至300,或甚至更优选地多至200种任何种类的分析物结合部分,如特异于任何基因或基因产物的寡核苷酸。
在一个进一步的方面,本发明涉及用于在样品中检测一种或多种甲状腺癌症标志物的方法,包括使用发明的组和检测样品中肿瘤标志物的存在或测量其发生量。所检测的标志物的发生率或模式能特异性鉴定这些标志物的存在,所述标志物能与癌症诊断相关或作为健康样品的参考,或简单地作为受试者的遗传调查。
优选地,样品包括细胞,优选地为哺乳类细胞,尤其优选地为人细胞,其能由活检组织或体液提供。具体而言肿瘤标志物的存在或量在例如细胞破碎之后在这些细胞中检测或测量。
所述方法包括通过RNA-表达分析的检测或测量,优选地通过微阵列或定量PCR,或蛋白质分析,优选地通过组织微阵列检测,蛋白质微阵列检测,mRNA微阵列检测,ELISA,多重测定法,免疫组化,或DNA分析,比较基因组杂交(CGH)-阵列或单核酸多态性(SNP)-分析。这些方法在本领域已知,并易于用于本发明的方法,如遗传标志物分析的广泛领域的实例。
在另一方面,本发明提供了用于在病人中诊断癌症的方法,包括提供病人的样品,优选地为病人的细胞样品,通过使用根据本发明的组测量检测肿瘤标志物信号而检测一种或多种肿瘤标志物,将所测量的肿瘤标志物的信号值与健康样品中肿瘤标志物的值进行比较,并且如果超过50%,优选地超过60%,更优选地超过70%,最优选地超过80%的值与健康样品的值相比的差异至少为测量方法的标准差,优选地两倍的标准差,甚至更优选地三倍的标准差,诊断为癌症。患病受试者和健康受试者样品之间的遗传表达的差异可以为任何种类,包括上调(例如原癌基因)或下调(例如肿瘤抑制基因)。有可能在健康样品中,基因不被表达,而在患病样品中发生表达。相反地,还可能是基因在患病样品中不表达而在健康样品中发生表达。
如果超过50%,优选地超过60%,更优选地超过70%,最优选地超过80%的样品的值与健康样品的值的差异为至少1.5倍,至少2倍,至少3倍或至少4倍,也可以诊断为癌症。通常肿瘤标志物表达产物上调或下调2-6倍,但60倍的差异也是可能的。
在另一方面,本发明涉及用于鉴定基本特异性标志物(例如在表1-6中给出的)的方法,优选地为基因或基因表达模式,包括:
●提供至少两种不同表达数据组的多种潜在的疾病特异性基因的基因表达数据,
●确定数据组的共同基因,
●将每个基因表达数据组标准化,优选地通过局部加权回归散点平滑法(lowess)或分位数标准化,
●将基因表达数据组合并为合并数据组,优选地将合并数据组标准化,并整合合并数据组,
●通过确定最近缩小重心(shrunken centroid)确定合并数据组的基因,其包括确定将基因分配到疾病的交叉证实的误差值及通过减少合并(优选地为标准化的)数据组成员数量而使误差值最小化,
其中减少的数据组的基因为特异于疾病的标志物。交叉证实能使用例如留一法(leave-one-out)。优选地,确定步骤(分类步骤)包括通过交叉-证实确定每个基因标准化的表达值与重心值的差异的最大阈值。然后具有低于阈值的标准化表达值的基因从减少(或缩小)组中移除,具有与重心相比大于阈值的值的基因对该疾病有特异性。通过缩小重心(shrunken centrois)方法进行的分类由例如Tibshirani等人(PNAS USA 99(10):105-114(2004)),Shen等人(Bioinformatics 22(22)(2006):2635-42)及Wang等人(Bioinformatics 23(8)(2007):972-9)公开,这些公开并入本文作为参考。
通过留出得自每个先前步骤的标志物能多次重复进行确定步骤。最近缩小重心法会产生特异于该疾病的进一步标志物的新的结果组。优选地,确定步骤重复2,3,4,5,6,7,8,9,10或更多次。根据合并数据组的大小,会给出进一步的特异性标志物。优选地,在每个结果上进行交叉证实。能重复进行确定直到交叉证实指出的误差值为例如低于50%,60%,70%或80%。在较低的值上,预期所有标志物都鉴定出来。
起始的基因表达数据组为原始的表达谱,例如得自多遗传微阵列分析的每个组。预期大多数所测量的基因不涉及疾病,且本发明的方法能够从至少两个,优选地至少三个,至少四个,至少五个,至少六个,至少七个或至少八个表达数据组中鉴定特征性的标志物基因。因此起始数据组的表达数据优选地包括至少两个不同微阵列数据组的数据,具体而言具有研究或平台特异性偏差。这些偏差是通过在测量表达数据过程中只使用一个特异设置而发生的,例如微阵列,其能显著的区别于其他数据组的设置。本发明具有的优点是在这些组的合并过程中,克服了这些测量偏差的问题。进一步,所得的(起始)基因表达数据是原始的,未处理的基因表达数据,即,在本发明的方法之前没有进行提炼或数据转换。
优选地,疾病为遗传障碍,优选地为具有基因表达改变的障碍,尤其优选地为癌症。具有基因表达改变的其他类型的障碍可以为例如病原体感染,具体而言为病毒(包括逆病毒)感染,辐射损伤和年龄相关障碍。
合并及整合合并的数据组的步骤去除了研究特异性偏差。在优选的实施方式中,此步骤通过逐步地,每步合并两个的基因表达数据组以及整合合并的数据而进行,优选地通过DWD(距离加权判别法,Distance Weighted Discrimination)。例如在3个数据组的情况下,首先将组1与组2合并,融合的组1+2与组3合并。整合例如可以包括计算整合的数据组的正常向量,接着计算将数据组(例如起始数据组的)数据值的聚类分开的超平面,以及减去数据组平均值,如DWD方法中的。原则上,任何移除偏差的数据整合方法能用于本发明的方法。
优选地至少一个,优选地两个,三个,四个,五个,六个,七个或八个所得的表达数据组包括至少10,优选地至少20,更优选地至少30,甚至更优选地至少40,至少50,至少70,至少100,至少120,至少140,至少160或甚至至少200种不同基因的数据。本发明的方法尤其适合于从大的数据组中进行过滤并鉴定其中的特征性标志物。所得的这些标志物的组还称为“分类物”。
鉴定癌症特异性标志物的此方法,以及因此特异于癌症的部分,例如寡核苷酸或抗体,还能用在诊断癌症的以上方法中。即,对应于用于诊断方法的部分的组的标志物是根据以上方法鉴定(也叫做“分类”)的,所述方法包括提炼及建立起始数据组的测量值的重心值。此模式然后能用于诊断癌症,如果病人样品值更接近于肿瘤标志物的聚类的重心值。因此,提供了用于在病人中进行癌症诊断的方法,包括提供来自病人的样品,优选地为细胞样品,通过使用根据本发明的组进行测量肿瘤标志物信号来检测一种或多种肿瘤标志物,通过以上提到的鉴定方法将肿瘤标志物的所测量信号值与癌症样品中的肿瘤标志物的值比较,并且如果对于至少50%,优选地至少60%,更优选地至少70%或甚至至少80%,最优选地为90%的组的标志物,病人样品的值的最近缩小重心在对用癌症样品鉴定出的肿瘤标志物的最近缩小重心的测量方法的标准差,优选地为两倍的标准差,甚至更优选地为三倍的标准差之内,诊断为癌症。
本发明进一步通过以下图和实例进行示例说明,而不特异性限制于其中。所有此处引用的参考文献并入本文作为参考。
图:
图1:DWD-整合前后的第一批两种主要的组分。根据图例,数据组用颜色编码,肿瘤实体由字母编码。
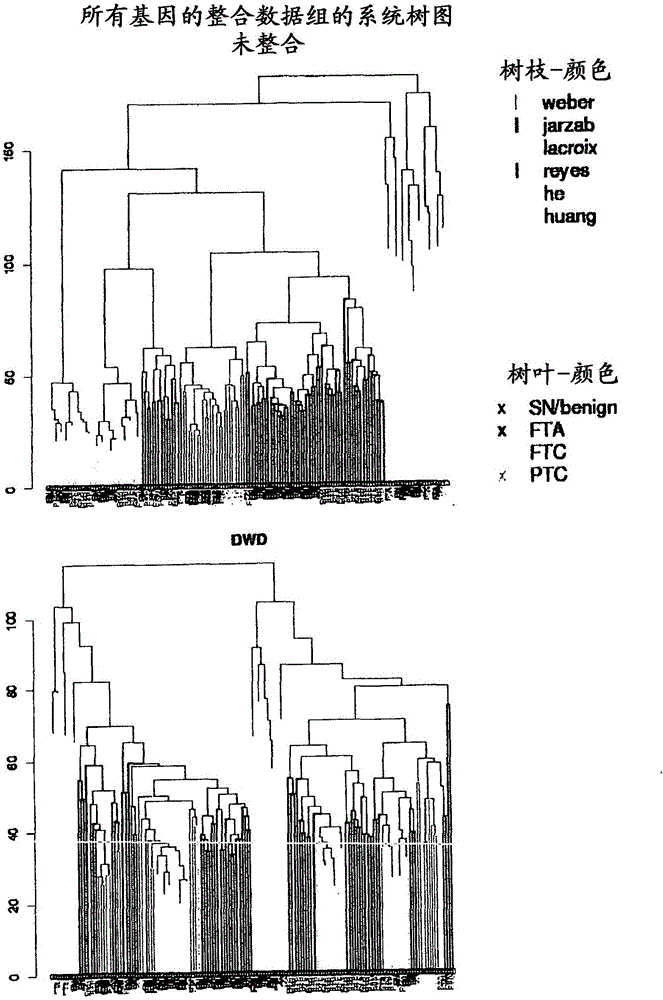
图2:所有基因的DWD整合数据的系统树图。系统树图的树枝颜色指示对应样品的数据组,树叶-标签的颜色指示了肿瘤实体。
图3:乳突状癌症和良性结节之间在四个不同数据组的差异只有一个基因(SERPINA1)
图4显示了来自表2的标志物的演绎组(分类物)的PTC分类中平均误差概率的图表。
图5显示了来自表4的标志物的演绎组(分类物)的FTC分类中平均误差概率的图表。
实施例
实施例1:数据组
数据组从网站下载或者来自公共知识库(GEO,ArrayExpress)。表7显示了在本研究中使用的数据组的总结(He等人,PNAS USA 102(52):19075-80(2005);Huang等人PNAS USA 98(26):15044-49(2001);Jarzab Cancer Res 65(4):1587-97(2005);Lacroix Am J Pathol 167(1):223-231(2005);J Clin Endocrinol Metab 90(5):2512-21(2005))。这里,使用了三种不同类别的非-癌症组织:对侧(contralateral,c.lat)用于与肿瘤样品配对的健康周围组织,其他疾病(other disease,o.d.)用于对其他疾病操作的甲状腺组织和SN(结节性甲状腺肿)用于良性甲状腺结节。对于所有随后分析,将这些合并为健康的。
表7:用于趋势分析的微阵列数据
实施例2:找到基因重叠
微阵列数据的任何趋势分析(meta-analysis)的第一步是找到分析中所用的所有微阵列平台所共享的基因的组。传统地,通过找到共同的UniGene识别号来评估重叠。然而这不考虑研究中基因的所有可能的剪接变异。例如,如果一个基因有2种剪接变体,其中之一种实验中有差异表达而另一种没有,以及如果一个平台包括只对该差异表达变体有特异性的寡核苷酸而另一个平台只有对另一种变体的寡核苷酸,则基于UniGene的匹配将融合测量不同事务的探针。
要克服这个问题,此处采用的手段仅仅融合了注解于RefSeq识别号的相同组的探针。为此,对于每个探针(组),通过Bioconductor注解包(hgu133a,hgu95a及hgu133plus2;在网站www.bioconductor.org上可得)或通过在NCBI数据库BLAST搜索序列下载所有匹配的RefSeq。然后,对于每个探针进行RefSeq的分类和和连结。这是阵列上所测量的实体的最精确的代表。如果一组RefSeq由阵列上多种探针所代表,则使用中值。在所有阵列上存在RefSeq的5707个不同组。
实施例3:预处理及数据整合
首先,如为每个平台所推荐的(局部加权回归散点平滑法用于双色实验,分位数标准化用于单色实验)(Bolstad等人Bioinformatics 19(2):185-193(2003);Smyth等人Methods 31(4):265-273(2003)),对每个数据组分别进行背景校正及标准化,然后将其融合并一起进行分位数标准化。尽管都进行了预处理,但显示不同微阵列平台上产生的数据或相同平台的不同产生的数据可能由于平台的特异性偏差而不可比(Eszlinger等人Clin Endocrinol Metab 91(5):1934-1942(2006))。这从融合的数据的主要组分分析中也很明显,如图1中所显示的。为了对这些偏差进行校正,开发了用于微阵列数据整合的方法。这些方法中的一种就是距离加权判别法(Distance Weighted Discrimination,DWD),其在别处有详细描述(Benito等人Bioinformatics 20(1):105-114(2004))。简言之,DWD将数据点投射到一类(数据组)的正常向量上——如通过修改的支持向量机(Support Vector Machine,SVM)计算的分离超平面并减去这类(数据组)平均值。因此,对于多类问题(要融合超过2个数据组),数据组需要顺序进行融合。对于6个数据组,这产生720种不同的可能性用于融合,不包括树结构的手段,例如,不是(((1+2)+3)+4),而是考虑((1+2)+(3+4))。此处应用的融合顺序是按更相似及更大的数据组应当首先合并,更不同的后合并,这一一般理念进行选择的。还值得注意的是,将样品加到DWD融合的数据组将改变整个数据组,就像将一个新数字加到许多数字的向量会改变其平均值一样。
通过DWD的数据整合在图1中阐述,其显示了在最先的两个重要组分上的数据整合方法的效应。在该分析中,DWD能够去除数据组之间的分离,如通过PC-作图及通过混合系统树图中树枝(见图2)所显示的。然而甚至在DWD-整合的数据组中,Lacroix数据仍然部分地与其他数据分离。这最可能是平台的缘故;lacroix-数据是来自非-Affymetrix平台的唯一数据。图2显示了各个整合的数据组的系统树图。而且,DWD整合似乎不妨碍在肿瘤实体间进行区分(见下列表8)。
实施例4:分类
对于探针选择,选择了分类及交叉-证实最近缩小重心方法(Tibshirani等人PNAS USA 99(10):105-114(2004))(在Bioconductor包pamr中施行)。由于如下几个原因选择了它:其允许多类别分类且一次运行特征选择、分类和交叉-证实。简言之,其使用不同收缩阈值(即,不同的基因数目)计算几个不同的可能分类物,并从交叉-证实中找到最好的阈值。如果超过一个阈值产生相同的交叉-证实结果,则挑选具有最小基因数量(最大阈值)的分类物。
实施例5:乳突状甲状腺癌(PTC)
首先,作为对于每项研究的质量测量,分开取出每个数据组(在DWD-整合之前)并进行pamr分类和留一法交叉-证实(leave-one-out cross-validation,loocv)。交叉-证实的结果几乎完美,只有单个样品分类错误。然而来自He数据组的分类物是例外,这些分类物中没有一个能应用于任何其他数据组。分类结果几乎不会比根据机会所预期的高。然而如果使用DWD-整合的数据(下文),分类物则已经适合地更好(见表8)。
表8:将来自一项研究的分类物应用于另一项研究的分类结果。数据整合之前(左)及DWD整合之后(右)
然后为完整的DWD-整合的数据组建立pamr-分类物并在留一法交叉-证实中进行证实。这鉴定了一个(!)基因分类物,其在loocv中正确分类了99%的样品。区分的基因为SERPINA1。图3显示了在DWD之前和之后PTC对SN的区分。能将多至422个基因加到分类物并仍然产生99%的精确度(来自loocv)。如果将SERPINA1-探针从分析中移除,能在loocv中再次以99%的精确度建立一个分类物(随后命名的分类物),这次使用的是一个9-基因签名(见表3)。移除这9个基因产生另一种9-基因分类物,其具有相似的表现(99%精确度),并且进一步产生一种具有99%精确度的11-基因分类物。这些进一步的分类物在例如用于PTC表1-3,5和6(上文)中给出。
然而在非-整合的数据上进行相同分析得到类似的结果。考虑到PCA的结果(图1),其中不同数据组所解释的方差显然比肿瘤实体所解释的方差大得多,可以想象由数据组引入的偏差有助于(或妨碍)分类。因此进行了研究-交叉证实,由此顺序地将一项研究从数据组中取出,分类物从剩余样品中建立并在消除的数据组上测试。在DWD-整合的数据中,预测精确度分别从分类物中留出He,Huang,Jarzab和Reyes为100,100,98及100%。对于非-整合的数据,结果是类似的(100,100,94及100%)。
表9:分类物2中的基因(留出SERPINA1后)
实施例6:滤泡性癌
对于FTC数据也进行了类似的分析,但交叉证实受到妨碍,因为得到数据非常受限。再一次地,为每个数据组建立分类物(Lacroix和Weber)。他们在25和3997个基因上达到了96%(Weber)及100%(Lacroix)的loocv-精确度。Lacroix-数据中的基因数量已经暗示了过度拟合,这通过其他数据组(分别为25和35%的精确度)的交叉-证实得到确定。而且,这两种分类物之间的基因-重叠很低(根据阈值,在0-10%之间)。然而如果使用DWD将这2个数据组合并,能建立147-基因分类物(上文表4),其能够正确鉴定样品(具有92%精确度)。
实施例7:讨论
本发明代表了迄今分析的甲状腺癌微阵列数据的最大同期组群。其利用新型的合并方法,使用用于微阵列数据整合和分类的最新算法。然而,微阵列数据的趋势分析仍然提出了一个挑战,主要因为单个微阵列研究的目标为至少部分不同的问题,且因此使用不同的实验设计。此外,迄今可得到甲状腺肿瘤微阵列数据的数量仍然相对较低(例如,相比于乳腺癌)。因此,在做趋势分析时被迫使用所有可得的数据,甚至如果病人同期群组代表着相当异质且潜在有偏差的群体。更特异地,难以得到对照材料(来自健康病人)的均质集合。这些通常取自因为其他甲状腺疾病做过手术的病人,其反过来很可能引起在微阵列上测量时基因表达发生变化。由于得到病人数据,例如年龄、性别、遗传背景等受限,均质的病人同期组群的产生进一步受到妨碍。
在做微阵列数据的趋势分析时,许多研究者将其手段基于了比较来自已发表的研究的基因列表上(Griffith等人,上文引用的)。这非常有用,因为能包括分析中的所有研究,且不限于可得到原始数据的研究。然而这些研究一般遵照了非常不同的分析策略,一些比其他更严密。作者如何作出基因列表这不在趋势分析者的控制之下。因此这些分析可能有偏差。
考虑到数据整合,根据原始的DWD文章,当每个数据组至少存在25-30个样品时DWD表现最好。在本研究中,6个数据组中有4个包括少于20个样品。在去除平台偏差上DWD仍然表现相对好(见表8)。
将来自一项研究的分类物应用于另一项研究时,DWD极大地改进了PCA的结果(图1),分级聚类(图2)以及分类精确度(表8)。在此基础上,令人惊奇地看到非-整合的数据在交叉证实研究中相比于DWD-整合的数据表现地同样好。其一个解释为任何研究-特异性偏差在评估更多研究时会变得不那么重要。鉴于研究偏差对一些基因的影响超过其他,由于研究-偏差引入的方差,受影响更多的基因更不可能经受pamr-阈值的界定。然而如上文显示的,有大量基因区分PTC和良性结节。只要这些基因中的一个(或几个)不受研究偏差的影响,它(它们)就能经受阈值界定,并且肿瘤实体间的区分仍然是可能的。
从图3中看到有个明显的矛盾:在DWD之前,PTC样品具有更高的SERPINA1表达,而DWD之后则相反。然而如材料和方法部分强调的,DWD从每个样品减去类的平均数。这简单地意味着在DWD之前,SERPINA1的研究偏差高于肿瘤类之间的表达差异。这还解释了为何在非-整合的数据中,SERPINA1不是工作很好的分类物。
一项近期的由Griffith等人进行的趋势分析及趋势回顾(Meta-Review)(上文引用的)总结了在甲状腺疾病背景中具有诊断潜能的基因。他们发表了在分析甲状腺疾病的超过一项高通量研究(微阵列,SAGE)中出现的基因列表,并应用了排序系统。在他们的分析中,SERPINA1打分为第三高,TFF3(留出SERPINA1时为分类物2的一部分)打分为第二。来自分类物2的九个基因中的四个出现在Griffith等人的列表中(LRP4,TFF3,DPP4和FABP4)。
这些列表的大多数从微阵列分析中产生。然而甚至当将分类物中基因与用独立技术(像cDNA文库产生)产生的基因列表相比时,有实质性的重叠。SERPINA1以及来自分类物2的九个基因中的四个(TFF3,DPP4,CHI3L1和LAMB3)出现在他们的列表中。
对于滤泡性甲状腺疾病的情况,建立强大的分类物更难。这主要因为得到数据受限。而且,这两个数据组在所使用平台方面非常不同;所有其他数据组都在不同产生的Affymetrix GeneChips微阵列上产生,而Lacroix数据在自定义的安捷伦(Agilent)平台上产生。然而表4的分类物(组)能够在loocv中正确鉴定大多数样品。
此处采用的趋势分析手段的效力由对于乳突状甲状腺癌和良性结节之间区别的99%的loocv-精确度(交叉证实研究中97.9%的加权平均精确度)所证实。迄今这在最大、最分散的数据组上实现了(来自4项不同研究的99个样品)。
一个样品被错误分类,尽管不可能正确地将此分析的样品分析对原始分析进行作图,错误分类的样品来自与原始分析中被错误分类的样品相同的组(PTC,证实组)。根据Jarzab等人,样品是异常值,因为其只包括≈20%肿瘤细胞。
- 还没有人留言评论。精彩留言会获得点赞!
- 普适型用于同时检测多种肿瘤标志物的微流控芯片装置的制造方法
- 男性适用的典型肿瘤标志物联合检测用微流控芯片装置的制造方法
- 女性适用的典型肿瘤标志物联合检测用微流控芯片装置的制造方法
- 一种检测口腔癌的标志物及其试剂盒的制造方法与工艺
- microRNA 标志物组及其在制备检测胃癌淋巴结转移试剂盒中的应用的制造方法与工艺
- 基于弹性蛋白酶荧光底物检测乳腺癌肿瘤标志物CA153的酶联免疫试剂盒制备方法与制造工艺
- 检测和评价力量训练效果的miRNA标志物或其组合及其应用的制造方法与工艺
- 一种最优超平面的构建方法、动态优化系统和构建装置与制造工艺
- 癌症标志物及癌症的判定方法与制造工艺
- 检测尿液中单羟酚类代谢物的试剂盒及其制备方法与制造工艺