一种红参中5种皂苷类成分的含量测定方法与流程
本发明涉及到一种红参中人参皂苷类成分的含量测定方法,属中药技术领域,具体的,涉及一种红参中人参皂苷Rg1、人参皂苷Re、人参皂苷Rf、人参皂苷Rb1和人参皂苷Ro五种成分同时测定的含量测定方法。
背景技术:
红参为五加科植物人参Panax ginseng C.A.Mey的栽培品经蒸制后的干燥根及根茎,秋季采挖,洗净,蒸制后,干燥,是药用历史悠久的传统名贵中药。人参被列为《神农本草经》三十六种上品之一,在历代中医药典籍中也均有记载。本经中称,人参:“主补五脏,安精神,定魂魄,止惊悸,除邪气,明目,开心益智,久服轻身延年”,是滋补养生保健的必备圣药,其功效长期以来被中医推崇备至。自古代起就有把采集到的野生人参,用煮、蒸、焯等方法进行加工处理,得到熟人参,这种加工方式逐渐演变成以后的红参。红参作为我国传统名贵中药,具有大补元气、复脉固脱、益气摄血的功效,长期以来深受广大人民群众的喜爱。人参皂苷类成分是红参的主要有效活性成分,是衡量红参内在质量优劣的重要标准,目前尚无红参中人参皂苷Rg1、人参皂苷Re、人参皂苷Rf、人参皂苷Rb1及人参皂苷Ro五种人参皂苷类成分同时测定的含量测定方法。
技术实现要素:
本发明的目的在于提供了一种红参中人参皂苷Rg1、人参皂苷Re、人参皂苷Rf、人参皂苷Rb1及人参皂苷Ro五种人参皂苷类成分同时测定的含量测定方法。
本发明含量测定方法采用超高效液相色谱法,该方法如下:
色谱条件与系统适用性试验色谱柱采用BEH C18色谱柱;以乙腈为流动相A,0.05%~0.10%磷酸为流动相B,进行梯度洗脱,洗脱梯度为:0~9分钟,流动相A:18%→20%;9~11分钟,流动相A:20%→28%;11~40分钟,流动相A保持28%;检测波长为203nm;流速0.4ml/min;
混合对照品溶液的制备
取人参皂苷Rg1对照品、人参皂苷Re对照品、人参皂苷Rf对照品、人参皂苷Rb1对照品、人参皂苷Ro对照品适量,精密称定,加甲醇制成每1ml含人参皂苷Rg1对照品80μg、人参皂苷Re对照品60μg、人参皂苷Rf对照品50μg、人参皂苷Rb1对照品150μg、人参皂苷Ro对照品100μg的混合溶液,即得;
供试品溶液的制备取红参粉末,过四号筛,0.8~1.0g,精密称定,置具塞锥形瓶中,精密加入80%甲醇25ml,密塞,称定重量,加热回流提取1~2小时,取出,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得;
测定法分别精密吸取对照品溶液与供试品溶液各1~2μl,注入液相色谱仪,测定,以外标法计算含量,即得。
本发明含量测定方法中,流动相B优选为0.05%磷酸;取样量优选为1.0g;测定法中分别精密吸取对照品溶液与供试品溶液的量优选为1μl,注入液相色谱仪。
本发明含量测定方法最优的技术方案为:
色谱条件与系统适用性试验色谱柱采用BEH C18色谱柱;以乙腈为流动相A,0.05%磷酸为流动相B,进行梯度洗脱,洗脱梯度为:0~9分钟,流动相A:18%→20%;9~11分钟,流动相A:20%→28%;11~40分钟,流动相A保持28%;检测波长为203nm;流速0.4ml/min;
混合对照品溶液的制备
取人参皂苷Rg1对照品、人参皂苷Re对照品、人参皂苷Rf对照品、人参皂苷Rb1对照品、人参皂苷Ro对照品适量,精密称定,加甲醇制成每1ml含人参皂苷Rg1对照品80μg、人参皂苷Re对照品60μg、人参皂苷Rf对照品50μg、人参皂苷Rb1对照品150μg、人参皂苷Ro对照品100μg的混合溶液,即得;
供试品溶液的制备取红参粉末,过四号筛,1.0g,精密称定,置具塞锥形瓶中,精密加入80%甲醇25ml,密塞,称定重量,加热回流提取1.5小时,取出,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得;
测定法分别精密吸取对照品溶液与供试品溶液各1μl,注入液相色谱仪,测定,以外标法计算含量,即得。
为了验证测定含量方法的稳定性、准确性、专属性及系统适应性,对方法进行了以下方法学验证实验,以确保该含量测定方法可以用于红参的质量控制。
1、仪器与材料
仪器:Dionex Ultimate 3000超高效液相色谱仪(双三元泵,DAD检测器),METTLER AE240电子天平,METTLER AE163电子天平。
对照品:人参皂苷Rg1(中检院,批号:110703~201529)、人参皂苷Re(中检院,批号:110754~201525)、人参皂苷Rf(中检院,批号:111719~201505)、人参皂苷Rb1(中检院,批号:110704~201424)、人参皂苷Ro(中检院,批号:111903~201102)购自中国食品药品检定研究院。
试剂:乙腈为色谱纯(Merck),水为超纯水(由MILLIPORE超纯水制水机自制),其它试剂均为分析纯。
2、方法与结果
2.1混合对照品溶液的制备
取人参皂苷Rg1对照品、人参皂苷Re对照品、人参皂苷Rf对照品、人参皂苷Rb1对照品、人参皂苷Ro对照品适量,精密称定,加甲醇制成每1ml含人参皂苷Rg1对照品80μg、人参皂苷Re对照品60μg、人参皂苷Rf对照品50μg、人参皂苷Rb1对照品150μg、人参皂苷Ro对照品100μg的混合溶液,即得。
2.2供试品溶液的制备
取本品粉末(过四号筛)约1g,精密称定,置具塞锥形瓶中,精密加入80%甲醇25ml,密塞,称定重量,加热回流提取1.5小时,取出,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
2.3色谱条件
色谱柱:WATERS ACQUITY UPLC BEH C18(2.1×100mm,1.7μm)
流动相:以乙腈为流动相A,0.05%磷酸水溶液为流动相B,进行梯度洗脱,梯度洗脱程序详见表1;流速0.4ml·min-1,柱温为40℃,检测波长为203nm,对照品溶液与供试品溶液进样量各1μl。
表1流动相梯度洗脱表
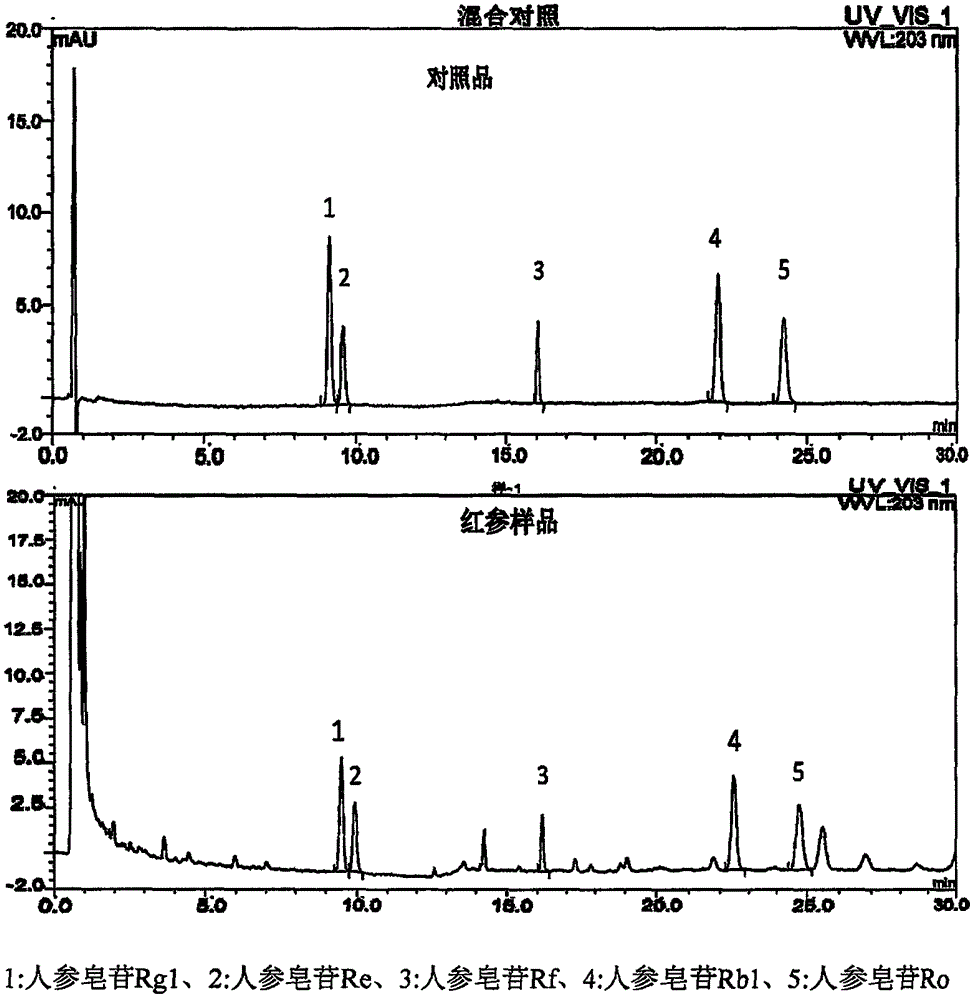
含量测定的色谱图见图1,可见各人参皂苷类成分分离良好,待测成分峰附近没有干扰峰,专属性较好。
2.4流动相选择
皂苷类物质紫外吸收非常弱,紫外检测器通常采用203nm波长检测,由于有机羧酸存在末端吸收现象,在203nm波长下基线噪音很大,干扰皂苷类含量测定,所以不选择甲酸、乙酸水溶液或甲酸盐、乙酸盐缓冲盐溶液作为流动相水相。实验摸索过程中考察了乙腈~水、乙腈~0.1%磷酸溶液、乙腈~0.05%磷酸溶液、乙腈~磷酸缓冲盐溶液等不同梯度的洗脱系统,发现用乙腈~水作为流动相时,人参皂苷Ro不能被洗脱分离;选用乙腈~磷酸缓冲盐溶液作为流动相时,人参皂苷Rb1与其他未知色谱峰不易分离,干扰准确测定含量。经反复试验,当采用乙腈~0.05%磷酸溶液作为流动相进行梯度洗脱时,基线平稳,5种人参皂苷分离度良好且峰形较佳。
2.5提取方式的选择
取同一批样品适量,选取80%甲醇作为提取溶剂,分别采用超声、回流两种提取方式进行试验,按拟定色谱条件测定。结果表明采用超声与回流两种提取方式,人参皂苷Rg1、人参皂苷Re、人参皂苷Rf、人参皂苷Ro的测定结果没有差别,而人参皂苷Rb1的测定结果回流明显高于超声,因此,选用回流作为本实验的提取方式(结果见表2)。
表2不同提取方式五种成分的含量测定结果
2.6提取溶剂的选择
取同一批样品适量,分别以甲醇、80%甲醇、60%甲醇、40%甲醇作为提取溶剂,回流处理1h。结果表明采用80%的甲醇作为提取溶剂五种成分的含量测定结果均较高,因此,选用80%的甲醇作为提取溶剂(结果见表3)。
表3不同提取溶剂五种成分的含量测定结果
2.7提取时间的选择
取同一批样品适量,回流提取时间分别为0.5、1、1.5、2h,按拟定色谱条件测定。结果表明回流1.5h能保证所有成分提取完全,故选择回流提取1.5h(结果见表4)。
表4不同提取时间四种成分的含量测定结果
2.8线性关系考察
精密量取人参皂苷Rg1(浓度依次为:0.01650mg/ml、0.06602mg/ml、0.082520mg/ml、0.1650mg/ml、0.3301mg/ml、0.8252mg/ml)、人参皂苷Re(浓度依次为:0.01323mg/ml、0.02646mg/ml、0.05293mg/ml、0.06616mg/ml、0.1323mg/ml、0.6616mg/ml)、人参皂苷Rf(浓度依次为:0.009568mg/ml、0.01914mg/ml、0.04784mg/ml、0.09568mg/ml、0.1914mg/ml、0.4784mg/ml)、人参皂苷Rb1(浓度依次为:0.02261mg/ml、0.09045mg/ml、0.1809mg/ml、0.4522mg/ml、0.9045mg/ml、2.261mg/ml)、人参皂苷Ro(浓度依次为:0.01202mg/ml、0.04809mg/ml、0.09619mg/ml、0.2405mg/ml、0.4809mg/ml、1.202mg/ml)1μl分别注入液相色谱仪,按“2.3”项下色谱条件测定,以对照品进样量(μg)为横坐标,峰面积积分值为纵坐标,绘制标准曲线。结果表明,人参皂苷Rg1在0.01650~0.8252μg之间呈良好的线性关系,回归方程为:y=644011x-593.85(r=0.9999);人参皂苷Re在0.01323~0.6616μg之间呈良好的线性关系,回归方程为:y=488436x+413.18(r=0.9999);人参皂苷Rf在0.009568~0.4784μg之间呈良好的线性关系,回归方程为:y=779269x~2959.3(r=0.9999),人参皂苷Rb1在0.02261~2.2612μg之间呈良好的线性关系,回归方程为:y=586991x+1538.7(r=0.9999),人参皂苷Ro在0.01202~1.202μg之间呈良好的线性关系,回归方程为:y=722156x~955.27(r=0.9999)。实验结果见表5。
表5红参五成分线性关系考察结果
2.9精密度试验
取混合对照品溶液,按“2.3”项下色谱条件测定,连续进样6次,记录色谱峰面积。结果显示各人参皂苷色谱峰面积R SD(n=6)均不大于2.0%。表明本方法的仪器精密度良好(结果见表6)。
表6精密度试验结果
2.10稳定性试验
取同一批样品适量,按“2.2”项下方法制备成供试品溶液,按“2.3”项下色谱条件测定,考察0、2、4、6、9、12、24h,结果显示各人参皂苷峰面积R SD(n=7)均不大于2.0%。结果表明,供试品溶液在24h内基本稳定(结果见表7)。
表7稳定性试验结果
2.11重复性试验
取同一批样品,研细,取0.5g、1.0g、1.5g各三份,精密称定,平行制成供试品溶液,按“2.3”项下色谱条件测定。结果表明,本方法重复性良好(结果见表8)。
表8重复性试验结果
2.12回收率试验
取已知含量的样品粉末9份,每份约0.5g,精密称定,每三份为一组,分别加入用80%甲醇溶液配制的低、中、高三个浓度的上述5种人参皂苷对照品溶液25ml,按供试品溶液制备项下方法制备供试品溶液。按“2.3”项下色谱条件测定,计算回收率。结果见表9~13。表明本方法回收率较好。
表9人参皂苷Rg1回收率试验结果
表10人参皂苷Re回收率试验结果
表11人参皂苷Rf回收率试验结果
表12人参皂苷Rb1回收率试验结果
表13人参皂苷Ro回收率试验结果
2.13样品测定
取收集到的红参药材、饮片共15批,按“2.2”项下方法制备成供试品溶液,按“2.3”项下色谱条件测定,计算含量,结果见表14。
表14红参样品含量测定结果
附图说明
图1红参中五种人参皂苷类成分含量测定色谱图;
具体实施方式
实施例1
色谱条件与系统适用性试验色谱柱采用BEH C18色谱柱;以乙腈为流动相A,0.05%磷酸为流动相B,进行梯度洗脱,洗脱梯度为:0~9分钟,流动相A:18%→20%;9~11分钟,流动相A:20%→28%;11~40分钟,流动相A保持28%;检测波长为203nm;流速0.4ml/min;
混合对照品溶液的制备
取人参皂苷Rg1对照品、人参皂苷Re对照品、人参皂苷Rf对照品、人参皂苷Rb1对照品、人参皂苷Ro对照品适量,精密称定,加甲醇制成每1ml含人参皂苷Rg1对照品80μg、人参皂苷Re对照品60μg、人参皂苷Rf对照品50μg、人参皂苷Rb1对照品150μg、人参皂苷Ro对照品100μg的混合溶液,即得;
供试品溶液的制备取红参粉末,过四号筛,1.0g,精密称定,置具塞锥形瓶中,精密加入80%甲醇25ml,密塞,称定重量,加热回流提取1.5小时,取出,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得;
测定法分别精密吸取对照品溶液与供试品溶液各1μl,注入液相色谱仪,测定,以外标法计算含量,即得。
实施例2
色谱条件与系统适用性试验色谱柱采用BEH C18色谱柱;以乙腈为流动相A,0.10%磷酸为流动相B,进行梯度洗脱,洗脱梯度为:0~9分钟,流动相A:18%→20%;9~11分钟,流动相A:20%→28%;11~40分钟,流动相A保持28%;检测波长为203nm;流速0.4ml/min;
混合对照品溶液的制备
取人参皂苷Rg1对照品、人参皂苷Re对照品、人参皂苷Rf对照品、人参皂苷Rb1对照品、人参皂苷Ro对照品适量,精密称定,加甲醇制成每1ml含人参皂苷Rg1对照品80μg、人参皂苷Re对照品60μg、人参皂苷Rf对照品50μg、人参皂苷Rb1对照品150μg、人参皂苷Ro对照品100μg的混合溶液,即得;
供试品溶液的制备取红参粉末,过四号筛,0.8g,精密称定,置具塞锥形瓶中,精密加入80%甲醇25ml,密塞,称定重量,加热回流提取1小时,取出,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得;
测定法分别精密吸取对照品溶液与供试品溶液各2μl,注入液相色谱仪,测定,以外标法计算含量,即得。
实施例3
色谱条件与系统适用性试验色谱柱采用BEH C18色谱柱;以乙腈为流动相A,0.05%磷酸为流动相B,进行梯度洗脱,洗脱梯度为:0~9分钟,流动相A:18%→20%;9~11分钟,流动相A:20%→28%;11~40分钟,流动相A保持28%;检测波长为203nm;流速0.4ml/min;
混合对照品溶液的制备
取人参皂苷Rg1对照品、人参皂苷Re对照品、人参皂苷Rf对照品、人参皂苷Rb1对照品、人参皂苷Ro对照品适量,精密称定,加甲醇制成每1ml含人参皂苷Rg1对照品80μg、人参皂苷Re对照品60μg、人参皂苷Rf对照品50μg、人参皂苷Rb1对照品150μg、人参皂苷Ro对照品100μg的混合溶液,即得;
供试品溶液的制备取红参粉末,过四号筛,1.0g,精密称定,置具塞锥形瓶中,精密加入80%甲醇25ml,密塞,称定重量,加热回流提取2小时,取出,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得;
测定法分别精密吸取对照品溶液与供试品溶液各2μl,注入液相色谱仪,测定,以外标法计算含量,即得。
上述实施例均按照药典进行方法学验证,结果显示方法准确可靠、灵敏、专属,各项指标均符合质量控制的要求。
- 还没有人留言评论。精彩留言会获得点赞!