用于组装来自一个或多个生物体的染色体段的方法、设备和计算机程序与流程
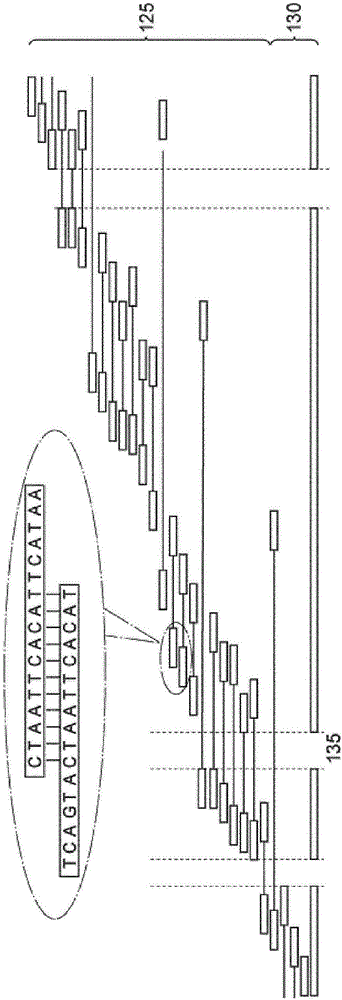
本发明总体上涉及基因组组装的领域。更具体地,本发明涉及用于使用与高通量测序结合的染色体构象捕获来组装一个或多个生物体的一个或多个基因组的方法、设备和计算机程序。发明背景微生物群落是维持环境稳定性和健康活生物体的基础。微生物物种最初是单独研究的,并且已经导致在诸如农业经济学、医学或消除污染的多种领域中开发了许多技术。由于技术的进步,现在有可能通过宏基因组学方法研究微生物群落的复杂性(例如超过100,000种不同的物种共存于一克土壤中,或数十亿微生物共存于人体内)。宏基因组学研究主要在于收集、测序和分析从直接从诸如皮肤、深海、肠、土壤、水等多样的环境中收集的微生物群落直接提取的遗传物质。DNA(脱氧核糖核酸)分子的数百万个随机段(其可以克隆到载体中)构成可以覆盖成千上万种不同物种的文库。对这样的文库的计算机(insilico)和实验分析导致发现新的基因和酶、新的网络和潜在的新物种(在地球上尚未发现的超过一千万种物种中的)。这种新方法不仅目前正在彻底改变我们对世界的理解,而且将最显著地在医学、能源和农业经济学领域中促进工业应用。许多公司已经开始挖掘这种未知的微生物多样性的巨大资源,同时一些机构和学院预测了关于这一新领域的未来的巨大前景。测序深度、读长长度和质量的快速改善已导致在集合种群内相对低丰度物种的基因组的表征。然而,由于在汇集重叠群和组装个体物种的大染色体区域的支架(scaffold)上遇到的困难,微生物群体的宏基因组分析仍然是有限的,从而损害其基因组中包含的信息的充分利用。基因组测序旨在确定DNA分子内核苷酸的顺序。DNA分子由两个生物聚合物链组成,这两个生物聚合物链彼此缠绕以形成双螺旋。该分子的每条链是被称为核苷酸的基本单元的聚合物。核苷酸由三个不同的部分组成:环状碱基(由鸟嘌呤-G、腺嘌呤-A、胸腺嘧啶-T或胞嘧啶-C制成),环状糖(脱氧核糖)和磷酸基团。在DNA分子中,核苷酸通过一个核苷酸的糖和下一个核苷酸的磷酸之间的共价键在链中彼此连接,导致交替的糖-磷酸骨架。根据碱基配对规则(A与T和C与G),氢将两个单独的多核苷酸链的含氮碱基键合以制备双链DNA。知道DNA序列(即四个环状碱基的连续顺序)对于生物研究以及在许多应用领域例如诊断、生物技术、法医生物学和生物系统学中是必需的。由于染色体通常包含数亿个核苷酸对,DNA测序仪的通量是许多实际应用(例如诊断)的关键因素。大规模DNA测序方法通常产生数百个或更少的碱基对的序列(即读长)。因此,在测序完整基因组之前,有必要将其剪切成更小的DNA段。这些片段被至少部分地单独测序以确定核苷酸的相应次序。那些DNA片段的仅小部分可以被测序(约100至200bp)。应当注意,可以使用导致两个读长的配对末端测序对DNA片段的末端进行测序。接下来,所获得的短序列必须重新组装以提供所研究的基因组的总体序列。根据众所周知的鸟枪测序方法,从生物体提取基因组并将其剪切成DNA的小段。接下来,对DNA片段进行测序,并基于重叠序列的完全相似性将所得的读长彼此重组以形成被称为重叠群的已知序列的DNA的部分。图1,包括图1a、1b和1c,示意性地说明了用于组合数百万个读长以形成重叠群并因此重组装片段的方法。如图1a所示,DNA片段100包含两条聚合物链110-1和110-2,其包含形成碱基对(bp)的核苷酸,例如序列对“ACTCTAATT”和“TGAGATTAA”。如上所述,DNA片段100只能从每端向内测序(箭头105-1和105-2)。DNA片段例如DNA片段100通常由结束于较粗的120-1和120-2(代表DNA片段的测序部分,即读长)的短线115代表。图1b示出了将DNA片段125组装到组装区域130中的过程。为此,分析读长并彼此比较以确定核苷酸的共同链。当两个DNA片段包含相同的核苷酸链时,它们作为该链在DNA片段中的相对位置的函数进行比对,如图所示。接下来,可以组装比对的DNA片段以形成重叠群,即DNA染色体段。应当注意,可以存在不对应于任何读长的部分,在组装区域中形成间隙,例如间隙135。换句话说,从DNA片段组装基因组作为核苷酸的重叠匹配序列的函数。图1c示出了重测序过程,根据该重测序过程,通过将短读长针对参照基因组145比对来重测序组装区域或重叠群140,如参照150-1和150-2所示。这种读长分析通常在计算机上递归地进行,以通过组装算法来组装片段。为了举例说明的目的,其可以通过已知名为IDBA-UD的算法进行(参见例如Bioinformatics,2012,Jun1;28(11):1420-8.doi:10.1093/bioinformatics/bts174.Epub2012,April11,IDBA-UD:adenovoassemblerforsingle-cellandmetagenomicsequencingdatawithhighlyunevendepth.PengY,LeungHC,YiuSM,ChinFY)。与实施组装算法的程序结合的鸟枪法测序方法可用于分析和重组装数百万个读长以获得通常包含高达30,000个碱基对的重叠群。然而,尽管该方法可能是有效的,但是由于基因组的大的重复部分在组装步骤期间引入模糊性并且使组装不完整,使得该方法具有限制性。为了提高组装效率,可以使用DNA的空间结构。实际上,与从DNA环产生的片段之间的观察到的接触结合的DNA空间结构的理论知识可以用于解决重叠群之间的冲突/模糊性或帮助缩小DNA序列中的间隙。例如,通过确定两个片段在空间上彼此接近,可以得出结论,这些片段沿着已从其获得这些片段的DNA纤维彼此接近。这主要是得自聚合物理学(染色体是半柔性的聚合物链,其经常在自身上成环用于小的基因组分离)。使用染色体构象捕获(3C)技术可以测定DNA的两个部分的空间接近性。3C技术及其随后的基因组变型(例如4C,5C和Hi-C)用于分析染色体的组构。图2,包括图2a至2f,示意性地示出了Hi-C技术的主要步骤。如图2a所示,第一步涉及将靠近在一起的DNA部分(例如,部分200-1和200-2)交联。这可以使用甲醛进行以用于将DNA的部分与蛋白质(例如蛋白质205)交联以及用于使蛋白质彼此交联。这导致使相接触的DNA部分交联。接下来,在第二步中,使用限制性酶将交联的DNA片段片段化。因此,如图2b所示,片段210-1和210-2分别由部分200-1和200-2的片段化产生。第三步旨在用产生平末端(以增加嵌合分子的比例)的修饰碱基(例如生物素)填充片段突出端。如图2c所示,片段210-2的末端用产生平末端并允许连接片段的免疫沉淀的材料215-1和215-2填充。在图2d所示的随后步骤中,连接DNA片段末端。接下来,逆转交联并纯化DNA。如图2e所示,剪切DNA片段并分离包含连接接合点的片段。最后,如图2f所示,将测序衔接子加入到DNA分子以产生可以测序的文库。测序这些片段允许鉴定由于染色体的空间结构而靠近在一起的DNA的部分。对测序的DNA段的空间结构的知识还可以用于鉴定属于一个或多个生物体的不同染色体的DNA的部分。实际上,可以认为DNA部分之间的接触可以用于聚类属于相同序列的DNA部分(通过考虑相接触的DNA部分属于相同的序列)。因此,为了对从单个或几个生物体得到的几个染色体进行从头测序,可以使用Hi-C类型方法产生第一文库,并使用鸟枪类型方法产生第二文库。然后,通过第二文库(即鸟枪类型)产生的重叠群可以作为第一文库(即HiC类型)包含的空间接触信息的函数进行聚类,从而导致将重叠群重组成更大的重叠群组。为了举例说明的目的,描述于题为“High-throughputgenomescaffoldingfrominvivoDNAinteractionfrequency”(N.Kaplan等人,NatureBiotechnology,vol.31,no.12,2013年11月24日)和“Chromosome-scalescaffoldingofdenovogenomeassembliesbasedonchromatininteractions”(J.Burton等人,NatureBiotechnology,vol.31,no.12,2013年11月3日)的文献中的方法主要基于用于测定DNA片段库的聚类步骤以及用于鉴定共线性的后续步骤。更精确地,在第一步中,将重叠群聚类成彼此之间具有高接触频率的组,并因此其可能属于相同的染色体。在第二步中,将每组内的重叠群相对于彼此重新排序,以使得连续的重叠群具有高接触频率,从而产生染色体支架。通过这些程序获得的最终支架可能反映单个染色体的真实线性结构。然而,这些方法存在几个缺点。首先,它们没有考虑重复性。特别地,在序列组装期间融合的重复区域不能使用这些方法解析。其次,在初始聚类步骤中的错误不能在支架化期间校正,导致最终的基因组组装品质强烈依赖于聚类准确度。第三,每种算法具有其自身的特定限制:第一种不试图定向支架中的重叠群,而第二种需要预先指定几个基因组特异性参数,包括染色体的确切数目,这将其应用限制到已经很好表征的基因组。最后,两种方法提出单基因组组装结果,而不论输入3C数据的品质和潜在模糊性,并且不提供关于其可靠性的全局或稳定的概率信息。发明概述面对这些约束和限制,本发明人提供了用于从单个生物体或从生物体的混合物高通量组装染色体段的方法、设备和计算机程序。本发明的广义目的是弥补上述现有技术的缺点。根据本发明的第一方面,提供了计算机方法,其用于基于代表至少一个文库的所有DNA片段的一组原始子序列组装代表至少一个生物体的至少一条染色体的至少一段的至少一个序列,所述至少一个文库包括包含所述至少一条染色体的连续核苷酸的链的DNA片段并且包括包含所述至少一条染色体的连续核苷酸的至少两条链的组合的DNA片段,所述方法包括以下步骤:-获得代表所述至少一条染色体的DNA区域之间的接触频率的第一值,所述第一值与代表相应DNA区域之间的距离的第二值相关联;和-迭代地执行以下步骤:-基于所述第一和第二值并基于将DNA区域之间的接触概率与相应DNA区域之间的距离相关联的理论模型更新基因组结构,更新的基因组结构代表所述至少一个生物体的至少一条染色体的至少一段的真实基因组结构;和-更新作为更新的基因组结构的函数的所述理论模型的参数。所要求保护的方法提供了一种有效的工具来校正和改善其基因组没有被充分表征的物种的基因组组装。特别地,本发明的方法使用共有相似胞腔区室(cellularcompartment)的染色体经历的频繁物理接触以组装物种中的个体染色体的大支架,而无需预先知道所预期的染色体的数量。此外,本发明的方法适用于来自不同生物体的细胞的混合物,从而允许宏基因组数据的去卷积。当进行本发明的方法时产生的读长还可以通过执行初步的从头组装步骤直接用于产生大的DNA重叠群。此外,本发明的方法使得能够在一个单一实验中产生存在于群体中的生物体的并且分箱(binning)在一起的大的重叠群。设想可以用这种方法研究非常小的集合种群。可以用本发明的技术分析许多种类的微生物组。此外,本发明提供了已知生物体的3D基因组结构的高通量表征,因为本发明使得可以一次评估数十个生物体的基因组结构(即序列)和3D基因组组构。旨在表征例如几个生物体中的染色体重排或这些基因组的3D组构的许多实验室或研究小组对这种方法感兴趣。本发明的方法提供了表征未知物种的3D基因组结构的方式。宏基因组方法显示盲法分析可以揭示物种的天然混合物中存在的未知物种的基因组序列和3D组构。染色体3D组构与代谢状态相关。因此,表征在混合物中共存的物种的3D组构揭示它们的代谢状态。这可以应用于解密这些物种相较彼此的生长状态,并鉴定集合种群的演变过程中的不同阶段(例如,应激、快速生长、静止)。在实施方案中,将两个DNA区域之间的距离测定为沿着预定路径的两个DNA区域之间的距离和/或两个DNA区域之间的空间距离的函数。在实施方案中,所述方法还包括将代表至少一个文库的所有DNA片段的原始子序列分成多个箱(bin)的步骤。在实施方案中,所述方法还包括产生多个基因组候选结构和计算每个所产生的候选基因组结构更接近于真实基因组结构的显式似然值(explicitlikelihoodvalue)的步骤。在实施方案中,产生多个基因组候选结构的步骤基于包括易位、缺失、倒置和重复中的至少一种变化的预定结构变化。在实施方案中,根据多重尝试Metropolis(multipletryMetropolis)类型的规则选择所生成的基因组候选结构之一作为相关联的似然值的函数。在实施方案中,基因组候选结构通过箱的结构变化来确定。在实施方案中,更新理论模型参数的步骤基于Gibbs采样类型的算法。在实施方案中,理论模型包括代表用于区分DNA区域之间的染色体内接触与DNA区域之间的染色体内和染色体间接触的阈值的至少一个参数。在实施方案中,理论模型包括代表用于区分DNA区域之间的染色体内接触或DNA区域之间的染色体内和染色体间接触与不同生物体之间的接触的阈值的至少一个参数。在实施方案中,所述方法还包括对至少一个文库的DNA片段进行聚类的步骤,其中每个聚簇与特定生物体相关,所述原始子序列对应于被处理用于基于聚簇测序的经聚类的DNA片段。在实施方案中,对文库的DNA片段进行聚类的步骤基于Louvain类型的算法。在实施方案中,所述方法还包括鉴定代表至少一个生物体的至少一条染色体的至少一段的至少一条序列中的至少一个DNA序列的步骤。在实施方案中,所述方法表征至少一个生物体的全局染色体组构,所述方法还包括推断所述至少一个生物体的代谢状态的步骤,所述全局染色体组构的特征在于相应基因组的三维组构。本发明的第二方面提供了用于鉴定生物样品中的真核细胞、原核细胞或微生物的基因组的方法,所述方法包括上述用于组装至少一个生物体的至少一条染色体的至少一段的方法的每一个步骤。在实施方案中,所述方法用于鉴定生物样品中的微生物的基因组,所述微生物是寄生虫、细菌、古细菌、真菌、酵母和病毒类型之一。那些细胞和微生物可以是致病性的(即对于植物或动物)或非致病性的。在更具体的实施方案中,生物样品含有或包含多于一种细胞或微生物种类。在实施方案中,用于组装至少一个生物体的至少一条染色体的至少一段的方法或用于鉴定基因组的方法还包括以下步骤:-使包含所述至少一条染色体的至少一段的所制备的生物样品的染色体段交联;-使用至少两种不同类型的限制性酶使交联的染色体片段化;和-对从片段化步骤得到的染色体片段进行测序。本发明的第三和第四方面提供了包含被配置用于执行上述方法的每个步骤的工具的装置,以及用于可编程装置的计算机程序产品,所述计算机程序产品包括用于当所述程序由可编程装置加载和执行时执行上述方法的每个步骤的指令。所要求保护的装置和计算机程序提供了校正和改善其基因组没有被充分表征的物种的基因组组装的有效工具。本发明的第五方面提供了用于组装至少一个生物体的至少一条染色体的至少一段的方法,所述方法包括以下步骤:-制备包含所述至少一条染色体的至少一段的生物样品;-使所制备的生物样品的染色体段交联;-使用至少两种不同类型的限制性酶使交联的染色体片段化;-对从片段化步骤得到的染色体片段进行测序;和-组装染色体的经测序的片段。所要求保护的方法提供了校正和改善其基因组没有被充分表征的物种的基因组组装的有效工具。在实施方案中,使用终浓度为3%的甲醛进行所制备的生物样品的染色体段的交联。在实施方案中,所述方法还包括经交联的染色体的机械裂解的步骤,所述机械裂解在使用至少两种不同类型的限制性酶的片段化之前进行。在优选的实施方案中,机械裂解是基于玻璃或陶瓷珠的。根据本发明的第六方面,提供了用于建立生物样品的病毒组(virome)和基因组之间的对应性的方法,所述方法包括以下步骤:-从所述生物样品中提取独立病毒颗粒群体;-基于权利要求1至17中任一项所述的方法鉴定提取的独立病毒颗粒群体的病毒基因组序列,所鉴定的病毒基因组序列形成所述病毒组;-基于权利要求1至17中任一项所述的方法鉴定其中已提取病毒颗粒群体的生物样品中的细菌、质粒和病毒基因组序列,以形成所述生物样品的基因组;和-基于物理接触建立所述生物样品的病毒组和基因组之间的对应性。本发明的方法使得可以确定病毒和细菌群体的组成,其平衡潜在地反映健康或环境条件。在实施方案中,所述方法还包括以下步骤:裂解提取的病毒颗粒群体的病毒,提取裂解的病毒的DNA,并从提取的DNA重建染色质。在本发明的优选实施方案中,病毒是噬菌体。由于本发明的一些部分可以以软件实现,所以本发明的一些部分可以实施为用于在任何合适的载体介质上提供给可编程装置的计算机可读代码。有形载体介质可以包括存储介质诸如软盘、CD-ROM、硬盘驱动器、磁带设备或固态存储设备等。瞬时载体介质可以包括信号诸如电信号、电子信号、光信号、声信号、磁信号或电磁信号,例如微波或RF信号。在实施方案中,计算机代码利用允许大矩阵数据的并行处理的图形处理单元(GPU)。可以使用图形技术实现DNA片段和组装过程的可视化,以允许在web界面上的可移植性。附图简述在检查附图和详细描述后,本发明的其它优势将变得明显。期望将任何额外的优势并入本文。现在将参考以下附图仅通过举例的方式描述本发明的实施方案,在附图中:图1,包括图1a、1b和1c,示意性地说明了组合数百万个读长以形成重叠群并因此重组装片段的过程;图2,包括图2a至2f,示出Hi-C技术的主要步骤;图3示意性地示出了根据本发明的实施方案的方法的主要步骤;图4示出了用于制备生物样品的步骤的实例;图5示出了从如参考图4所述制备的生物样品构建Meta3C文库的实例;图6示出了使用从不同生物体的混合物获得的Meta3C文库用于确定基因组组构和基因组支架的第一个实例;图7示意性地示出了对应于Meta3C文库的片段的一组原始子序列和从这些原始子序列衍生的一组重叠群;图8示出了使用从不同生物体的混合物获得的Meta3C文库用于确定基因组组构和基因组支架的第二个实施例;图9示出了用于确定一个或几个不同生物体的基因组组构和支架的GRAAL算法的步骤;图10,包括图10a至10e,示出了图9中呈现的某些步骤;图11,包括图11a和11b,示出了可以在DNA区域之间的接触概率与这些DNA区域之间的距离之间建立的关系;图12,包括图12a、12b和12c,示出了在GRAAL算法的三个不同迭代(t=0,t=501,和t=4,500)下生物样品的DNA区域和相应基因组结构的DNA区域之间的接触阵列的实例;图13是示出其中可以至少部分地实现本发明的实施方案的处理设备的组件的框图;图14至17示出了用于确定生物样品中的噬菌体群体和细菌群体的组成的应用的实例;和图18示出了图14至17中呈现的步骤。本发明的实施方案的详细描述根据本发明的实施方案,共有相似胞腔区室的染色体经历的频繁物理接触(可以通过染色体构象的宏基因组捕获(Meta3C)实验来测量)可以用于组装存在于集合种群中的基因组的较大支架。不仅Meta3C文库可允许组装大的DNA区域(基于它们的接触频率),而且可以通过执行初步的从头组装步骤直接使用读长来产生它们。图3示意性地示出了根据本发明的实施例的方法的主要步骤。如图所示,第一步骤(步骤300)旨在制备可用于构建Meta3C文库(步骤305)的生物样品。Meta3C文库由一组DNA片段组成。这些片段的组成通过其末端的配对末端测序测定。如果方案不涉及嵌合片段的富集步骤,这些序列可用于从头组装较长的重叠群。这些读长还用于确定生物样品中存在的基因组的结构和相关参数(步骤310)。分别参考图4和5描述步骤300和305,而通过参考图6和8(其代表本发明的实施方案的两个实例)描述步骤310。图4示出了用于制备生物样品的步骤(即,图3中的步骤300)的实例。稀释(例如在150mlPBS(磷酸缓冲盐水)中)生物样品(例如30mg湿材料(例如来自河流沉积物的湿物质))(步骤400)。应当注意,对于固体样品、对于需要小心操作的样品和/或对于量太小而不能以大体积处理的样品,不需要这样的稀释步骤。接下来,对其进行化学处理,以便通过交联蛋白质和DNA的部分来冻结全局染色质网络(步骤405)。这种处理可以用新鲜甲醛(例如3%终浓度)在室温下进行30分钟然后在4℃下进行30分钟来实现。在下一步骤中,停止交联反应过程(步骤410)。为了举例说明的目的,甲醛可以用0.25M甘氨酸的终浓度在室温下进行5分钟然后在4℃进行15分钟来淬灭。接下来,固定的细胞通常通过离心收集,洗涤(例如通过使用50mLPBS),再次收集(例如通过离心),并在干冰中冷冻并储存在-80℃直到使用(步骤415)。必须注意,旨在获得具有DNA片段的一致交联的初始生物样品的步骤(步骤405)是必需的。因此,特别注意产生固定的细胞以获得足够量的交联片段。图5示出了从如上所述制备的生物样品构建Meta3C文库(图3中的步骤305)的实例。如所示的,第一步骤(步骤500)旨在对冷冻的细胞沉淀进行解冻。为了举例说明的目的,这可以在冰上在30分钟内完成。接下来,将解冻的细胞重悬(步骤505)。这可以在650μl1×TEpH8的终体积中进行。在随后的步骤中,裂解样品细胞(步骤510)。这可以在均质器下已知的设备中进行,根据该设备,使用玻璃或陶瓷珠机械地裂解细胞。为了举例说明的目的,可以以6,700转/秒的速度每60秒进行三个20秒的循环裂解样品细胞。接下来,合并裂解的细胞并用10%cc浓度的SDS(十二烷基硫酸钠)处理,以获得例如0.5%cc的最终SDS浓度,并将合并的细胞例如在室温孵育15分钟(步骤515)。然后将经处理的裂解细胞分配在几个管中(步骤520)。然后用不同的限制性酶将存在于这些细胞内的DNA分子片段化(步骤525-1至525-3,一般称为525)。由于原始生物样品通常包含不同生物体的细胞,可以使用几种类型的限制性酶以使得不同类型的染色体组成(关于GC和AT碱基对的比例)可以被片段化成平均大小不超过预定阈值(通常为500至1,000个碱基对)的小段。此阈值对于不呈现嵌合读长的富集步骤的Meta3C文库是特别重要的。应注意,虽然在图5中仅呈现三种不同的限制性酶(步骤525-1至525-3),但是可以组合使用或在独立过程中使用任何数量的限制性酶。如步骤525中所示,将裂解的细胞置于含有消化混合物(例如1XNEB1缓冲液(10mMBis-Tris-丙烷-HCl,10mMMgCl2,1mMDTT,在25℃pH7.0,NewEnglandBiolabs),1%TritonX-100和预定类型的100U限制性酶)的管中。将裂解的细胞和消化混合物的掺合物在相应的酶活性温度(例如37℃)下孵育(步骤530)预定的时间段,例如3小时。根据具体实施方案,选择所使用的限制性酶以便识别四种碱基对组合。因此,用于制备生物样品的交联步骤需要比通常用于制备DNA片段文库的浓度和时间更高的浓度和更长的时间。可以使用识别六种碱基对组合的酶或其它的酶,但是文库的构建需要富集步骤,例如在DNA片段的边缘处掺入生物素化的碱基。接下来,在随后的步骤中,将含有裂解细胞和消化混合物的掺合物的管离心(步骤535),例如以16,000转/分钟的转速离心20分钟。离心后,移除漂浮物(上清液),并将沉积在管底部上的材料重悬浮(步骤540),例如在500μl水中。然后合并管(例如3mL),并将连接混合物加入到所获得的混合物中(步骤545)。为了举例说明的目的,连接混合物可以基于NEB(NewEnglandBiolabs)连接缓冲液(例如1.6mL)、BSA(牛血清白蛋白)(例如10mg/mL,160μl)、250UT4DNA连接酶和水以获得16mL的体积。将混合物例如在16℃下孵育4小时。接下来,停止反应并反转交联(去交联步骤550)。这可以通过向混合物中加入EDTA(乙二胺四乙酸)(例如100mM终浓度)和蛋白酶K(例如4mg),并通过将混合物例如在65℃下孵育12小时来实现。然后提取DNA片段(步骤560),例如通过异丙醇沉淀、酚氯仿沉淀,和通过乙醇沉淀(例如用Tris10mM(例如60μL)恢复),并且施加RNA酶处理以除去RNA分子。最后,合并所有管,从而得到Meta3C文库,其使用例如由Bio-Rad开发的称为QuantityOne的应用(QuantityOne和Bio-Rad是商标)在凝胶上定量。通过进行参考图4和5描述的步骤获得的Meta3C文库通常包含几百个或更少的碱基对的读长。可以通过使用标准组装算法(例如IDBA-UD)将文库的重叠读长一起拼接到更大的连续序列(重叠群)中。应注意,通过进行参考图4和5描述的步骤而获得的Meta3C文库(没有嵌合序列的富集步骤)主要包括对应于存在于从其延伸文库的生物样品中的染色体的片段的DNA分子(即,染色体的连续核苷酸的链)。这些片段被称为鸟枪类型的DNA片段,因为它们与在鸟枪测序文库中恢复的DNA片段相似。这些鸟枪DNA片段占文库的片段的约80%。Meta3C文库还包含在交联步骤期间捕获在一起的两个序列的组合(即连续核苷酸的至少两个不同链的组合)的DNA分子,也称为嵌合分子。这两个序列越频繁地彼此接近(图4的步骤405),则它们将越频繁地被捕获在一起。这些序列可以沿着相同的DNA纤维(染色体)相距很远地定位或定位在分开的染色体上(即嵌合分子是相同染色体或至少两个不同染色体的连续核苷酸的至少两个不同链的组合)。DNA片段之间的这些长距离相互作用代表约20%的文库分子。与存在于Hi-C文库中的嵌合分子相反,当进行这些嵌合分子的富集步骤时,Meta3C文库中的嵌合分子不包含在组合的序列之间的生物素。因此,Meta3C文库包含“鸟枪”和“嵌合”形式两者的DNA分子,因此可用于在单个实验中产生重叠群并对其支架化(scaffold)。应当注意,当“鸟枪”分子数量较少时,MetaHiC文库可以用于对通过其它方法获得的重叠群进行支架化。图6示出了使用从不同生物体的混合物获得的Meta3C文库用于确定基因组组构和基因组支架的第一个实例。图6中所示的算法基于从Meta3C库获得的标记为600的一组原始子序列。原始子序列是来自Meta3C文库中存在的DNA片段的配对末端测序的读长对。第一步骤旨在将Meta3C文库的DNA片段组装成被称为重叠群的更大序列(步骤605)。该步骤可以通过标准组装算法(例如IDBA-UD)来执行。作为结果,获得了一组重叠群,标记为610。然后使用构成不同重叠群的读长的配对末端信息来产生所有重叠群相互之间的接触网络。为此,将从Meta3C文库的测序获得的配对末端读长在作为步骤605的结果获得的重叠群610上使用读长比对应用进行比对(步骤615)。应当记住,读长比对应用(也称为比对器)使得可以沿着用作参考的较大的DNA序列例如的染色体(或染色体的一部分)比对(即,定位)读长。为了举例说明的目的,被称为Bowtie2的应用是比对器,并且可以用于执行步骤615)。包含在DNA分子中的配对末端信息通过揭示哪些重叠群与哪些其它重叠群接触来确定重叠群之间的接触网络,标记为620。所发现的重叠群之间的这些相互作用的频率揭示了界定其的连接的强度。例如,转向图7,可以确定读长的接触网络。图7示意性地示出了对应于Meta3C文库的片段的原始子序列的集合700和从这些原始子序列衍生(例如根据参考图6描述的步骤605和615)的重叠群的集合705。如图所示,每个原始子序列末端代表经测序的数据(这些经测序的数据是读长)。当原始子序列共有相同的读长时组合原始子序列以形成重叠群。为了举例说明的目的,仅呈现了对应于二十四个DNA片段的二十四个原始子序列,每个子序列包含读长的配对末端。例如,子序列700-1包括配对末端读长R7和R8。类似地,仅呈现了三个重叠群(705-1,705-2和705-3)。如图所示,每个重叠群基于子序列的子集,并且包括读长的配对末端以及内部读长(已被用于组装目的)。例如,重叠群705-1包含配对末端读长R0和R3。如上所述,诸如IDBA-UD的标准组装算法的使用使得能够通过比较读长形成重叠群来组装DNA片段。因此,子序列700-2可以与子序列700-3组合以形成重叠群705-2(即重叠群R4-R5-R6)。为了举例说明的目的,可以假设重叠群705-1和705-2属于相同的染色体,而重叠群705-3属于不同的染色体。在这种情况下,R3和R4之间的连接很弱,从而削弱这些片段的组装。接触网络的构建揭示了包含在由远距离的非连续序列构成的嵌合分子中的信息项。例如,鉴于重叠群705-1和705-2中的读长R1和R5之间的距离,子序列700-4被认为与嵌合分子相关。如图7所示,可以从子序列集合705建立的连接遵循连续的结构直到位置R4/R5和R6/R7。在这些位置,出现间隙,因为由于某些原因,没有发现配对末端信息来连接重叠群。然而,可以从在组装中未使用的嵌合分子的配对末端信息项中鉴定长距离信息项。这样的信息项揭示了在组装过程中不明显的重叠群之间的连接。长距离信息项可以用于将重叠群彼此定位在它们最可能的取向中。通过确定重叠群中每个子序列末端的位置(这样的距离可以表达为多个碱基对的函数或多个限制性片段的函数)和通过将多个接触(或接触频率)与这些位置中的每一个相关联,可以从子序列集合700和从所获得的重叠群集合705构建接触阵列。为此,所有重叠群以线性结构组装,重叠群的读长序列形成如下文所示的接触阵列的排和列的参考:R0R1R2R3R4R5R6R7R8R9R00200000000R12050020000R20504000100R30040000000R40000030000R60200303000R60000030000R70010000030R80000000301R90000000010为了实现接触阵列,将后者的每个单元在初始化步骤中设置为零。接下来,选择子序列集合的子序列(通常一个接一个),并且对于每个子序列,对应于子序列的一端的读长的位置在组装的重叠群的线性结构中确定,以确定第一坐标(a)。类似地,在组装的重叠群的线性结构中确定对应于子序列的另一端的读长位置,以确定第二坐标(b)。然后,第一和第二坐标用于鉴定接触阵列的两个对称单元((a,b)和(b,a))的内容增加1。或者,可以实现仅一半的接触阵列,另一半与第一个重复。回到图6,后续的步骤涉及聚类Meta3C文库的DNA片段(步骤625)。这可以例如使用Louvain类型的算法来完成。应该记住,Louvain算法是用于鉴定复杂网络中的聚簇或群落的简单而有效的方法。它基于一般定义,根据该定义,如果节点对是相同群落的成员,则它们更有可能被连接,并且如果它们不共有任何群落,则不太可能被连接。因此,通过知道节点之间的连接和连接的数量,可以鉴定代表群落的聚簇。在整个重叠群集合上通过使用其整体接触网络应用Louvain算法以将它们聚类为在它们之间显示优先接触频率的重叠群子集,允许DNA片段根据它们所属的生物体聚类,如标记630所示。因此,在步骤625中确定的聚簇被用于将Meta3C文库的大多数DNA片段分类为重叠群的库,其随后可以单独处理以更精确地确定这些生物体的每一个的支架,以及(最终地)它们的基因组组构。如标记635所提示的,信息项可以与每个确定的聚类相关联。接下来,在随后的步骤中,执行也被称为GRAAL(用于来自3D接触数据的基因组(Re)-组装评估似然性)的3D支架算法(步骤640),以确定每种生物体的基因组组构和支架,分别如标记645和650所示的。GRAAL是将虚拟重排(或结构变化)迭代地应用于DNA片段的初始集合的算法。该方法基于概率公式,其基于接触网络数据和将预期的接触频率与基因组结构相关联的先前(数据无关)假设计算提出的基因组结构的似然性。这些假设利用了这样的事实:预测的和观察到的染色体内接触频率与基因座之间的基因组分离密切相关,通常遵循近似幂律关系并且对于大基因组分离显示平顶(plateau),其中频率变得与染色体间接触频率相当。用于GRAAL算法的初始化的子序列从一组重叠群或从参考基因组产生。将它们有利地分成小至一个限制性片段的限制性片段的规则箱。如参考图9至图11所描述的,在每次迭代时,GRAAL算法挑选新的基因组箱并针对从测量的接触频率采样的N个箱扫描整个基因组。然后,考虑V个不同的虚拟结构变化(包括易位、删除、倒置和重复),计算相对于这些潜在领域的一组基因组。然后,在该组基因组的局部似然性景观上对候选箱进行采样,并且保留最可能的结构之一用于下一次迭代。基因组中每个箱的位置(不仅是预组装的重叠群)由GRAAL算法独立测试几次,从而允许减轻组装错误和鉴定非常小的结构变化。图8示出了使用获自不同生物体的混合物的Meta3C文库用于在单个分析步骤中使用GRAAL确定基因组组构和基因组支架的第二个实例。如图所示,步骤600'至620'分别类似于参考图6描述的步骤600至620。这些步骤旨在提供一组重叠群和接触网络信息。在随后的步骤(步骤800)中,所获得的一组重叠群和接触网络信息项用于直接确定(即,不执行聚类步骤,诸如参考图6中的步骤625所描述的Louvain算法)存在于用于产生经处理的Meta3C文库的生物样品中的每种不同生物体的基因组组构和支架,分别标记为645'和650'。如图8所示,然后可以使用基因组组构和支架来表征用于产生经处理的Meta3C文库的生物样品中存在的生物体(步骤805)。确定基因组组构和支架的步骤800优选地基于参考图6简要介绍并且参考图9至11更详细地描述的GRAAL算法,其中基因组结构的理论模型适于区分存在于所分析的生物样品中的不同生物体的染色体。如图所示,GRAAL算法使用理论模型810来迭代地确定一组染色体的结构815,该结构在每次迭代中用于更新理论模型810。这里要注意,即使在图6中未示出任何理论模型和染色体结构,在步骤640执行的GRAAL算法也基于使用理论模型和染色体结构,其相互作用以收敛于解。GRAAL算法是蒙特卡罗马尔科夫链(MonteCarloMarkovChain)(MCMC)类型的算法。其目的在于探索可能在所观察的数据(即Meta3C文库)的起源处的基因组结构的分布。属于相同生物体或不属于相同生物体的基因组结构的元件之间的接触频率在计算机化理论模型中建模。分析基因组结构的分布使得能够确定存在于用于产生经处理的Meta3C文库的生物样品中的不同生物体的可能数量,并确定每个生物体的基因组。确定存在于生物样品中的不同生物体的可能数量是基于频率接触变化的。对于给定的Meta3C文库,以D表示,GRAAL算法试图估计与数据一致的一维基因组结构G的整体概率分布p(G|D)。该算法基于使用贝叶斯规则p(G|D)∝p(D|G)p(G)的概率方法。假设在没有数据的情况下,所有结构G具有相等的概率(平坦先验(flatprior)),贝叶斯规则可以如下地减少:p(G|D)∝p(D|G)。p(D|G)的计算需要模型来定量预测染色体内接触和染色体间接触,其对于给定的G被称为顺式(cis-)和反式(trans)染色体接触矩阵M。假定顺式接触概率Pc取决于基因组分离s作为幂律(接着是平顶):对于s≤s0,Pc(s)=Ptsbs0-b,并且对于s>s0s>s0,PcO)=pt,根据限制在核中的染色体的理论预测的和测量的行为。应当注意,对于不同的生物体或染色体已报道不同的b和s0值。考虑到没有关于染色体邻域的先前信息和相对于顺式接触的反式接触频率的相对弱势,还假定反式接触以每单位基因组长度平方的均匀概率Pt发生。通过GRAAL算法估计参数ε=(b,s0,Pt),也称为冗余参数(nuisanceparameter)。此外,假设测量的接触矩阵M的计数服从泊松分布,即P(Mi,j=k)=λi,jke-λi,j/k!(keN))其中箱(i,j)的接触概率λi,j分别由反式或顺式接触的Pt或Pc给出。这些假设一起指定允许计算给出Meta3C文库(数据集D)的任何基因组结构G的似然性p(D|G,ε)的概率模型p(G,ε|D)。蒙特卡罗马尔可夫链算法(即Gibbs采样器)用于顺序地产生冗余参数和基因组结构。通过经典的Metropolis算法,用基因组结构的变化迭代地、交替地更新冗余参数。多重尝试Metropolis采样器(其生成从初始基因组结构G0开始的基因组结构Gt(t=1,2....Nt)的有序序列)用于产生基因组结构。给定当前基因组结构Gt,通过应用虚拟结构变化来计算N个新结构的随机集合,所述虚拟结构变化可以包括例如插入、缺失、重复、倒置、易位或这些的同时组合。对于每个候选结构变化,如上所述计算新结构的似然性,并且对基因组结构的空间执行局部随机优化以确定下一个结构Gt+1。用MetropolisTryMetropolis规则指定的概率接受或拒绝新的基因组。与结构变化的统一选择相反,此程序允许结构概率密度的计算高效采样。最后,在丢弃老化期(burn-inperiod)后,使用马尔可夫链样本来估计联合概率分布(G,b,s0)。图9示出了用于确定一个或几个不同生物体的基因组组构和支架的GRAAL算法的步骤。图9结合图10进行描述,图10包括图10a至10e,其示出了图9中呈现的某些步骤。如图10a所示,来自不同生物体的细胞混合物1000(例如细胞1005-1,1005-2和1005-3)(其包含与相应的一个生物体相关的基因组)用于产生Meta3c文库(代表DNA片段的子序列可以组合以形成一组重叠群,例如图10b中所示的重叠群1010-1至1010-4)。为了举例说明的目的,重叠群1010-1代表细胞1005-1的DNA片段的组装,重叠群1010-2代表细胞1005-2的DNA片段的组装,重叠群1010-3代表细胞1005-3的DNA片段的组装,以及重叠群1010-4代表细胞1005-1和1005-3的DNA片段的组装。换句话说,重叠群1010-4源自细胞1005-1的DNA部分与细胞1005-3的DNA部分之间的接触。不按照在DNA部分中这样的存在的重叠群1010-4也被称为嵌合读长对。回到图9,第一个所示步骤涉及对先前由标准组装算法确定的重叠群进行分箱(步骤900)。根据步骤900,将每个重叠群分成代表重叠群的部分的箱。箱的长度可以由用户调整。有利地测定为读长之间的接触数量的函数和/或读长之间的接触频率的函数。根据一个具体实施方案,所有箱的箱长度大致相同,并且其最小尺寸设置为等于限制性片段(即由限制性酶的片段化步骤产生的DNA片段)。在执行步骤900之后获得的对应于Meta3C文库的整个DNA片段集合的一组n个箱可以在以下关系式下表达:B={b1,b2,...,bi,...,bn}。箱的实例在图10c中示出。为了举例说明的目的,重叠群1010-3被分至包括箱1010'-31至1010'-3n的箱1010'-3的集合中。接下来,回到图9,构建箱接触阵列(步骤905)。它被构建为接触网络的函数,也就是说作为重叠群读长之间的接触频率的函数。虽然接触网络限定了重叠群i的位置a和重叠群j的位置b之间的接触,但箱接触阵列呈现重叠群i的箱a和重叠群j的箱b之间的接触的数量。如上所述,这些接触中约80%来自沿着DNA纤维相邻的DNA片段,并且这些接触中约20%来自沿着DNA纤维不相邻但在交联步骤(当构建Meta3C文库时)期间在空间上彼此接近的DNA片段。在随后的步骤中,初始化GRAAL算法的参数(步骤910)。在这些参数中,在执行GRAAL算法期间修改的基因组结构Gt被初始化至参考结构G0(即t=0)。这样的参考结构可以是例如已组装的基因组、部分组装的基因组或一组重叠群或箱。为了举例说明的目的,将基因组的初始结构设定为一组重叠群,即包含嵌合读长对的一组组装的DNA片段。待初始化的GRAAL算法的另一参数代表基因组结构的理论模型P(s)的参数ε=(b,s0,Pt),在初始化步骤期间该理论模型被表示为Pε0(s)。根据特定实施方案,模型P(s)将两个箱之间的接触概率与这两个箱之间的距离相关联(该距离通常被测定为沿着DNA纤维的这两个箱之间的距离的函数和/或空间距离的函数(例如,如果在两个箱之间存在接触,则距离等于一,否则为零))。参数(b,s0,Pt)可以被认为代表比例因子,和两个阈值R0和R1,其表征相同染色体的两个箱、不同生物体之一的两条染色体的两个箱和两个不同的生物体的两条染色体的两个箱之间的连接。图10b中示出了这种理论模型的示例。待初始化的另一个参数(表示为l)代表GRAAL算法必须运行的迭代次数或循环次数。它代表基因组结构经受的突变的数目,也就是说可以带给基因组结构的结构变化的数目。为了举例的目的,参数l可以设置为4,500。待初始化的另一参数(表示为V)代表基因组结构所经受的结构变化的类型。这种结构变化包括例如箱的易位、缺失、倒置和重复和/或这些结构变化的组合。待初始化的另一参数(表示为θ)代表当生成候选基因组结构时对于给定箱必须考虑的相邻箱的数量。为了示例的目的,参数θ可以被设置为10。在已经初始化这些参数之后,索引i被设置为包括在1和集合B={b1,b2,...,bi,...,bn}的箱的数量n之间的值并且选择箱i(步骤915)。索引i的值被选择为使得每个箱至少被选择一次且优选地被选择相同的次数(例如10)。图10d示出了一组箱的示例,其中选择一个被标记为1010'-rs的箱(箱i)。回到图9,在已经选择箱i之后,在从分箱步骤900得到的n个箱中选择一组θ箱(步骤920)。根据特定实施方案,θ个选择的箱被选择为箱i的θ个可能的领域。这可以通过随机选择与具有箱i的大量接触相关联的θ箱来完成。表示为箱(j)的θ个选择的箱中的第一个被鉴定为待处理的。接下来,然后使用所选择的箱i和所鉴定的箱j来生成表示为Gm,j的一组候选基因组结构(步骤925)。这些候选基因组结构被确定为箱i和j的函数和基因组结构所经受的结构变化的预定类型V的函数。对于所生成的表示为Gm,j的基因组结构中的每一个,计算相应的基因组结构是真实基因组结构的似然值(步骤930)。似然值通常通过将箱之间的预测接触与给出当前模型参数εt的该结构的所观察到的数据进行比较来获得。在已经生成针对鉴定的箱j的候选基因组结构并且计算这些候选基因组结构中的每一个的似然值之后,将参数θ递减1(步骤935),并且执行测试以确定其值是否等于零(步骤940)。如果变量θ的值不为零,则重复后四个步骤(步骤920至935)以鉴定θ个选定箱的集合中的不同箱j,以生成候选基因组结构的新集合作为新鉴定的箱j的函数,以及计算所生成的每个候选基因组结构的似然值。相反,如果变量θ的值等于零,也就是说,在针对所选择的箱i和针对θ个所选择的箱中的每一个生成候选基因组结构之后,生成的候选基因组结构之一被选择为下一个基因组结构Gt+1(步骤945),即Gt+1=Gm,j。根据具体实施方案,所产生的候选基因组结构之一的选择基于多重尝试Metropolis规则(MTM)。应当注意,可以使用其它标准,特别是用于优化计算性能的较少限制性标准(例如,基本随机优化)。图10e示出了针对所选择的箱i和针对θ个所选择的箱中的每一个生成候选基因组结构的步骤。接下来,回到图9,基于观察的数据和基因组结构Gt+1来更新基因组结构的理论模型P(s)的参数εt+1(b,s0,Pt)(步骤950)。为了举例说明的目的,这可以通过应用经典Gibbs采样器算法来完成。在更新了基因组结构的理论模型P(s)的参数的值之后,变量l递减1(步骤955),并且执行测试以确定其值是否等于零(步骤960)。如果变量l的值不为零,则θ的值被初始化为代表必须考虑的相邻箱的数量的值(步骤965),并且该算法分支到步骤915用于选择新的箱i和随后用新选择的箱i重复步骤920至950。如上所述,从900产生的箱集合中的每个箱优选地被选择一次,并且有利地选择大约十次。相反,如果变量l的值等于零,则算法停止。图11,包括图11a和11b,示出了可以在DNA区域之间的接触概率与这些DNA区域之间的距离之间建立的关系。更精确地,图11a示出了分别表征属于不同生物体的相同一个的两个不同染色体的DNA区域之间和属于两个不同生物体的两个不同染色体的DNA区域之间的接触概率的两个距离阈值R0和R1的示例。根据具体实施方案,DNA区域之间的距离是通常取决于沿着一对DNA链的距离(其对于相同染色体的DNA区域特别相关)和空间距离(其对于不同染色体的DNA区域是更相关的)的特定函数。图11b是代表两个DNA区域之间的距离(表示为横坐标)与这两个区域之间的接触概率(表示为纵坐标)之间的关系的图。为了举例说明的目的,认为染色体按每个生物体分组并且实际上彼此连接成单链,沿着该单链可以建立DNA区域之间的距离。如图所示,如果两个DNA区域属于相同染色体,则这两个DNA区域之间的距离通常小于R0,并且这两个DNA区域之间的接触概率高,如曲线1100的部分所示。类似地,如果两个DNA区域属于单个生物体的两个不同染色体,则这两个DNA区域之间的距离通常包含在R0和R1之间,并且这两个DNA区域之间的接触概率是中等的,如曲线1105的部分所示。最后,如果两个DNA区域属于两个不同生物体的两个不同染色体,则这两个DNA区域之间的距离通常大于R1,并且这两个DNA区域之间的接触概率低,如曲线1110的部分所示。换句话说,理论模型旨在预测作为两个基因组位置的函数的接触频率。所示的理论模型包括三个水平。第一水平针对染色体内接触,而第二水平针对染色体内和染色体间接触,第三水平针对不同生物体之间的接触。第一和第二水平由R0值分开,而第二和第三水平由R1值分开。第一水平基于聚合物的物理学。在执行GRAAL算法期间调整参数R0和R1,同时研究相应的分布。当然,根据例如对特定生物体特异的性质,可以使用多于两个阈值R0和R1。根据特定实施方案,DNA区域x和y之间的接触概率f与这些DNA区域之间的距离d之间的关系可以如下表达:其中C1和C2是常数值。应注意,可以如下修改关系f以考虑区室之间的接触的变化性:更一般地,这种关系可以被修改以适应特定需要。为了举例说明的目的,用于预测生物体内和生物体间接触的常数C1和C2可以被其它常数替代和/或完善,以考虑到亚组织例如共生和区室内重组。图12,包括图12a、12b和12c,示出了在GRAAL算法的三个不同迭代(t=0,t=501,和t=4,500)下生物样品的DNA片段和相应基因组结构的DNA片段之间的半接触阵列的实例。半接触阵列代表作为它们在染色体结构中的估计位置的函数排序的两个箱之间的接触的数量。当启动GRAAL算法时,箱被伪随机地排序,如图12a所示,其中接触阵列1200-0和染色体结构1205-0不代表任何特征模式。在501次迭代之后,一些聚簇开始出现在接触阵列1200-501上,并且一些图案可在染色体结构1205-501上鉴定,如图12b所示。最后,在4,500次迭代之后,在接触阵列1200-4500上可以清楚地识别聚簇,并且染色体结构1205-4500显示结构化的信息项,如图12c所示。图13示意性地示出了被配置用于实现本发明的至少一部分的至少一个实施方案(例如参考图6、8和9描述的算法中的一个或几个)的处理设备1300。处理设备1300可以是设备诸如微计算机、工作站或高度并行计算机。设备1300包括通信总线1313,其中优选地连接有:-中央处理单元1311,诸如微处理器,表示为CPU;-只读存储器1307,表示为ROM,用于存储用于实现本发明的计算机程序;-表示为RAM的随机存取存储器1312,用于存储本发明实施方案的方法的可执行代码以及适于记录实现根据本发明的实施方案测定基因组结构的方法所需的变量和参数的寄存器;和-连接到通信网络1303的通信接口1302,可以在其上传送要处理的数字数据。可选地,装置1300还可以包括以下部件:-诸如硬盘的数据存储工具1304,用于存储用于实现本发明的一个或多个实施方案的方法的计算机程序和在实现本发明的一个或多个实施方案期间使用或产生的数据;-用于磁盘1306的磁盘驱动器1305,磁盘驱动器适于从磁盘1306读取数据或将数据写入所述磁盘;-屏幕1309,用于通过键盘1310或任何其它指示工具显示数据和/或用作与用户的图形界面。通信总线在包括在装置1300中或连接到装置1300的各种元件之间提供通信和互操作性。总线的呈现不是限制性的,并且特别地,中央处理单元是可操作的以直接地或通过装置1300的另一元件向装置1300的任何元件传送指令。磁盘1306可以由任何信息介质代替,例如高密度磁盘(CD-ROM)(可重写的或不可重写的)、ZIP盘或存储卡,并且一般来说,信息存储意指可由微型计算机或微处理器读取、集成或不集成到装置中,可能是可移动的并且适于存储一个或多个程序,所述程序的执行使得能够实现对数字图像序列进行编码的方法和/或解码根据发明待实施的比特流的方法。可执行代码可以存储在只读存储器1307中、硬盘1304上或可移动数字介质上,例如如前所述的磁盘1306。根据变型,可以借助于通信网络1303经由接口1302接收程序的可执行代码,以便在被执行之前存储在装置1300的存储工具之一(诸如硬盘1304)中。中央处理单元1311适于控制和指导根据本发明的一个或多个程序的软件代码的指令或部分、存储在前述存储工具之一中的指令的执行。在通电时,存储在非易失性存储器(例如硬盘1304上)或只读存储器1307中的一个或多个程序被传送到随机存取存储器1312中,随机存取存储器1312随后包含所述一个或多个程序的可执行代码,以及用于存储实现本发明所需的变量和参数的寄存器。在该实施方案中,装置是使用软件来实现本发明的可编程装置。然而,可选地,本发明可以以硬件(例如,以专用集成电路或ASIC的形式)来实现。已经显示染色体3D组构与代谢状态相关。因此,通过表征在混合物中共存的许多物种的3D组构,可以揭示它们的代谢状态。这可以应用于通过彼此比较来破译这些物种的“生长”状态,以及鉴定在集合种群的进化期间的不同阶段(例如应激、快速生长、静止等)。此外,这种方法可以推动基于所涉及的不同物种(在诊断实验中潜在的令人感兴趣的)的染色体之间的相互作用的共生和寄生事件的鉴定。实际上,可以认为,更多的膜围绕遗传物质,则存在于不同细胞器中的物质在3C重连步骤期间相互作用的可能性越小。换句话说,实验的“噪声”将是所涉及的DNA片段的空间接近性的指示。例如,当两个基因组在空间上更远时,寄生虫的基因组在细胞内增殖阶段期间比在感染发生之前更多地与宿主基因组相互作用(例如在巨噬细胞的液泡中)。可能需要开发新的交联剂以增加信噪比(朝向噪声)。该应用对于开发在侵入性过程之后的诊断工具可能是重要的。所公开的用于组装代表一个或多个生物体的染色体段的序列的方法可以用于鉴定生物样品中的真核细胞、原核细胞或微生物的基因组。特别地,所公开的方法可以用于鉴定生物样品中的微生物的基因组,所述微生物是寄生虫、细菌、古细菌、真菌、酵母和病毒类型之一。那些细胞和微生物可以是致病的性(即对于植物或动物)或非致病性的。在更具体的实施方案中,生物样品含有或包含多于一种细胞或微生物种类。应当理解,如果GRAAL算法可以与Meta3C文库一起使用,则可以使用其它类型的DNA片段文库,例如Hi-C文库或鸟枪文库与Hi-C文库的组合。更一般地,GRALL算法可以与代表DNA片段并且包括这些DNA片段中的一些之间的接触或相邻性的信息项的数据一起使用。此外,应当注意,通过Meta3C方法研究基因组的物理特征在分析微生物的复杂群体(即病毒,特别是噬菌体(即在细菌内感染和复制的病毒))方面具有重要的潜力,。应当记住,噬菌体颗粒以非常高的量存在于天然群体中,并且它们的溶解性允许它们通过离心容易地与微生物分离。由于噬菌体在细菌宿主内繁殖,噬菌体颗粒的整个群体来自微生物的子集。重要的是表征噬菌体和细菌群体的组成,因为这个平衡潜在地反映健康或环境条件。然而,难以解释噬菌体颗粒群体来自哪个细菌菌株,因为噬菌体基因组难以在经典宏基因组测序中鉴定。同时,可溶性级分中的噬菌体颗粒的基因组序列由于这些序列的复杂的嵌合模式而不容易获得。到目前为止,对样品中自然群体内的噬菌体序列的良好理解是不可能的。上文所述的Meta3C解决方案可以解决这些问题。图14至17示出了这种应用的示例。图14示出了一般原理,而图15至17给出了图14所示的步骤的细节。在获得生物样品(步骤1400)后,将游离或独立的噬菌体颗粒群体与样品的其余部分分离(步骤1405)。然后,鉴定包含在这些颗粒内的噬菌体基因组序列(形成样品或群体的病毒组)(步骤1410)。这样的鉴定步骤可基于如上所述的Meta3C过程。平行地(或之前或之后),鉴定其中已除去噬菌体颗粒的剩余微生物的所有基因组(质粒,病毒)(步骤1420)。参考图15更详细地描述表示为1425的步骤1405、1410和1415。如图15所示,第一步骤(步骤1500)旨在将生物样品分成两个等分部分,一个部分用于鉴定噬菌体颗粒的序列,另一个部分用于鉴定在除去噬菌体颗粒后微生物的所有基因组、质粒和病毒序列。接下来,从第一部分的天然样品中分离噬菌体颗粒(步骤1505)。将这些颗粒裂解,并在溶液中回收噬菌体DNA分子(步骤1510)。这个DNA是裸露的。在随后的步骤中,在体外重构染色质,例如使用标准商业试剂盒(步骤1515)。自然地,其它选择是可能的,例如设计交联化学品,在宽距离范围上桥接裸露的DNA区域,或使用组蛋白和组蛋白样蛋白质提取物。然后使用Meta3C处理染色质,Meta3C数据使得可以产生存在于含有病毒颗粒的可溶性级分中的噬菌体序列的目录(步骤1520)。伴随地,如上所述通过Meta3C处理减去了噬菌体颗粒的天然样品(步骤1525)。表征该群落中存在的微生物的基因组。然后,基于如上所述的3D接触中的富集,质粒和病毒以及噬菌体(均是在游离体下的,即染色体外或整合的,即原噬菌体形式)也被表征并分配至它们各自的宿主细胞(图14中的步骤1420)。将在这些生物体内鉴定的噬菌体序列与存在于可溶性级分中的噬菌体的序列进行比较。这揭示了哪些细菌产生在可溶性级分中发现的不同噬菌体。在图16中示出了执行表示为1530的步骤1505和1510的示例,并且在图17中示出了执行表示为1535的步骤1515和1520的示例。图18示出了图14至17所示的步骤。如图所示,生物样品1800包含细胞,例如细胞1805-1、1805-2和1805-3,以及通常表示为1810或1810'的噬菌体颗粒。每个细胞通常包括细菌和质粒基因组以及噬菌体基因组。为了举例说明的目的,细胞1805-1包含噬菌体颗粒1810-1、噬菌体基因组1835-1和细菌基因组1815-1。表示为游离或独立噬菌体的这些噬菌体中的一些可以繁殖为存在于细菌外部的噬菌体颗粒,如标记1810'所示的。如上所述并且如标记1820所示的,从生物样品中提取细菌外的噬菌体颗粒,以便对其噬菌体基因组进行测序,从而形成噬菌体组(phageome)。伴随地,如标记1825所示,对其中噬菌体颗粒已被除去的生物样品的细菌、质粒和噬菌体基因组进行测序。然后,如标记1830所示,建立噬菌体组和基因组之间的对应性。自然地,为了满足局部和特定的要求,本领域技术人员可以将许多修改和变化应用于上述解决方案,然而,所有这些修改和变化都包括在由权利要求书所限定的本发明的保护范围内。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1