一种基于蒙特卡洛交叉验证的NIRS异常样本的检测方法与流程
本发明涉及一种异常样本的检测方法,尤其涉及一种基于蒙特卡洛交叉验证的NIRS异常样本的检测方法。
背景技术:
近红外光谱(Near Infrared Spectrum,NIRS)分析技术是一种依据样本成分特征进行分析的无损检测技术,近年来被广泛应用于农作物、石油化工、医药等多个领域。利用NIRS分析技术对样本进行定量或定性分析时,首先根据建模样本建立参比值和光谱数据之间的关系模型,然后将模型用于未知样本的光谱数据分析。在此过程中,建模样本的准确性直接决定所建模型的优劣。但通常情况下,由于样本本身及采集技术手段等原因,异常样本的出现是不可避免的,而异常样本的存在可能会严重影响模型的预测能力和模型评价的准确性。
样本异常通常由光谱数据误差和参比值误差导致。其中光谱数据的误差主要是由光谱扫描前的样本处理、光谱仪器本身、实验环境引起的;参比值误差主要是由实时操作不当、测量方法有误或者数据统计错误造成的。
传统的异常样本的判别都是以模型预测精度为依据,所以模型不稳定或者预测精度不高,容易造成异常样本的误判或多判。通常,在进行异常样本判别之前对模型稳定性的考察,以及判别之后异常样本是否被完全、准确识别是该研究中的两个易疏忽点。同时多数经典的判别异常样本的方法只是单方面地针对化学值或光谱数据,即使将判断化学值异常与判断光谱数据异常两种方法综合考虑,依旧会忽视化学值与光谱数据之间的联系,导致边界数据样本的误删。常见地,马氏距离法通常只是用来判别光谱数据的异常,而Cook距离法仅可用来识别样本化学值的异常。
本发明中使用组外判别法判定最佳主成分时使用的样本与预测集无关,所以当校正集确定时,主成分数也同样被确定了,在之后的使用中无需再次进行判定,从而简化了模型复杂程度,同时也提高了模型的稳健性;在进行MCCV实验前,随机划分校正集与预测集建立多个O-PLSR模型考察模型的SEP值分布范围,以此判别模型的稳定性;MCCV判别异常样本的原理是通过建立大量的模型,统计建模数据,根据异常样本的统计规律异于正常样本来判别样本异常,该方法充分考虑到化学值与光谱值之间的内在联系,实现了更加全面、准确的判别异常样本;通过剔除异常样本并重复MCCV实验来验证异常样本是否完全剔除。
技术实现要素:
本发明的目的在于克服现有技术的缺点与不足,提供了一种基于蒙特卡洛交叉验证的NIRS异常样本的检测方法,实现对光谱数据误差和参比值误差造成的异常样本的判别。
本发明的目的通过以下技术方案实现:1、一种基于蒙特卡洛交叉验证的NIRS异常样本的检测方法,包括如下步骤:
步骤1:使用组外判定法确定预处理过的光谱数据的最佳主成分,并结合偏最小二乘回归PLSR方法建模得到优化后的PLSR预测模型O-PLSR;
步骤2:判断O-PLSR模型的稳定性;
步骤3:使用蒙特卡洛交叉验证法MCCV随机建立多个O-PLSR模型,识别出强影响点;
步骤4:利用二审判别法判别强影响点中的异常值;
步骤5:剔除异常样本重复MCCV实验,验证异常样本是否剔除完全。
进一步的,所述步骤1具体为:
1.1使用优化后的KS算法选取与校正集和预测集均无交叉的样本M个,仅用来确定最佳主成分;
1.2再次采用优化后的KS算法将剩余的样本按照3:1的比例划分为校正集和预测集;
1.3对所有样本数据进行预处理,其中预处理方法为移动窗口拟和多项式平滑、标准变量变换以及一阶导数之一或其任意组合;
1.4用校正集内所有的样本点以及h个主成分拟合成一个回归方程,并将M个样本点的光谱数据代入拟合好的回归方程,得到M个样本的预测值,将预测值和参考值进行比较,计算得到预测标准差SEP,其中h=1,2,3,...h;
1.5统计使用不同主成分h拟合的回归方程对应求得的SEP值,选取最小的SEP值所对应的主成分为最佳主成分;
1.6使用最佳主成分建立PLSR模型,即O-PLSR模型。
进一步的,所述步骤3具体为:
3.1随机分配校正集与预测集建立多个O-PLSR模型;
3.2计算每个模型的SEP值,并依照这些值从小到大的规则对模型排序;
3.3按照排序后的模型统计每个样本在校正集中出现的累计频率;
3.4将累计频率高于设定比例或者低于另一设定比例的样本定为强影响点。
进一步的,所述步骤4具体为:
4.1利用随机法划分样本集合,得到校正集样本,该校正集样本包含强影响点,使用组外的M个样本作为预测集;
4.2分别将强影响点从校正集中剔除,建立O-PLSR模型,并分别考察模型对校正集与预测集的预测效果,对模型有贡献的强影响点视为正常样本,而对模型无贡献并且产生坏的影响的强影响点判别为异常样本。
进一步的,所述步骤5具体为:
5.1剔除异常样本再次进行MCCV实验,确定强影响点;
5.2对强影响点进行二审判别,检验异常样本是否剔除完全。
进一步的,所述优化后的KS算法是先将依据化学值大小排序后的样本分成N个区间,再对每个子区间使用KS算法划分校正集与预测集。
进一步的,所述累计频率定义为每个样本在模型校正集中出现概率,计算公式如下:
其中i指样本的序号,j为排序后模型的序号,如果样本i出现在模型j的校正集中,fij为1,否则为0,J是所统计的模型数量。
本发明能够更加全面、准确的判别异常样本,从而保证样本完全剔除。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面以小麦样本蛋白质化学值和光谱数据为实施例的研究对象,并将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还可以根据这些附图获得其他的附图。
图1:预测均方根误差变化分布图;
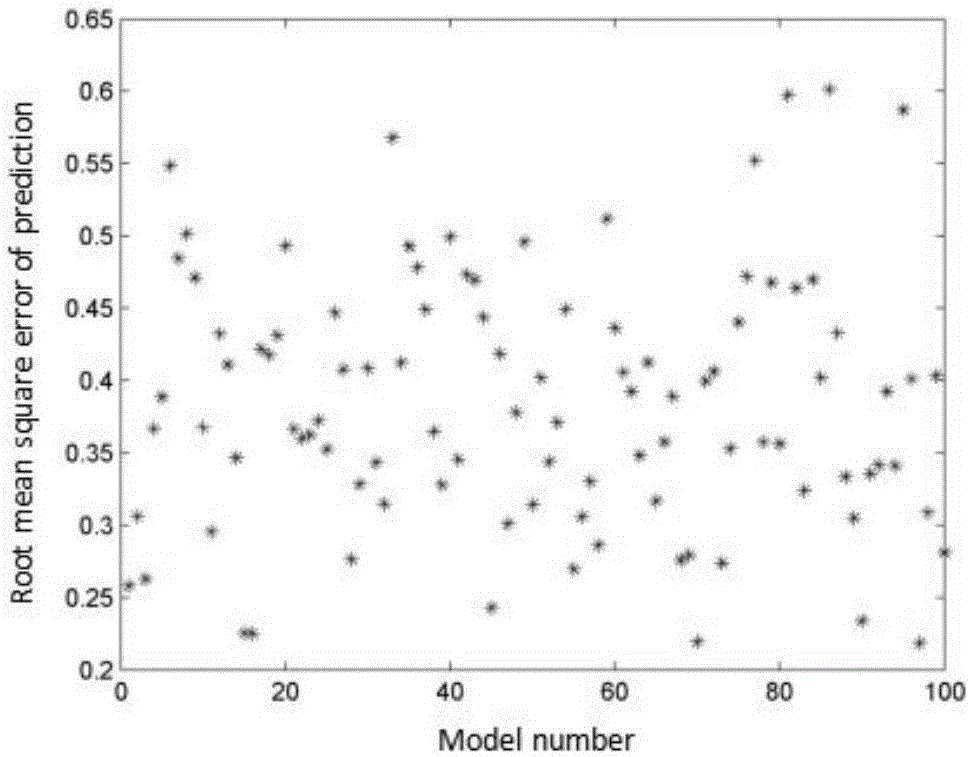
图2:100次模型的预测均方根误差分布图;
图3:各样本的累计频率变化图;
图4:前450个模型各样本的累计频率分布图;
图5:验证样本是否剔除完全实验的前450个模型各样本的累计频率分布图;
具体实施方式
下面将结合本发明实施例中的附图对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
小麦样本来源于2011年长江上游、长江中下游、东北、西北6个地区,共116个。分别对这些样本进行近红外扫描以及凯氏定氮法测量,得到116组小麦蛋白质的化学值与光谱数据。
本发明提供一种基于蒙特卡洛交叉验证的NIRS异常样本的检测方法,包括如下步骤:
步骤1:使用组外判定法确定预处理过的光谱数据的最佳主成分,并结合偏最小二乘回归PLSR方法建模得到优化后的PLSR预测模型O-PLSR;
步骤2:判断O-PLSR模型的稳定性;
步骤3:使用蒙特卡洛交叉验证法MCCV随机建立多个O-PLSR模型,识别出强影响点;
步骤4:利用二审判别法判别强影响点中的异常值;
步骤5:剔除异常样本重复MCCV实验,验证异常样本是否剔除完全。
其中步骤1中为建立一个稳健的O-PLSR模型,首先使用组外判别法确定建模的最佳主成分,具体如下:
(1)使用优化后的KS算法选取与校正集和预测集均无交叉的样本10个,仅用来确定最佳主成分;
(2)再次采用优化后的KS算法将剩下的106个小麦样本按照3:1的比例划分为校正集和预测集(即校正集中有80个样本);
(3)分别使用S-G平滑、SNV、1D三种预处理方法对116个样本数据进行预处理,比较这几种方法的预测结果,最终选用S-G平滑、SNV、1D同时对样本数据进行数学校正;
(4)用校正集内所有的样本点以及h个主成分拟合成一个回归方程,并将10个样本点的光谱数据代入拟合好的回归方程,得到10个样本的蛋白质预测值,将预测值和参考值进行比较,计算其SEP值;其中h=1,2,3...,100
(5)统计使用不同主成分h拟合的回归方程对应求得的SEP值,最终选取最小的SEP所对应的主成分为最佳主成分。
使用最佳主成分建立PLSR模型,即O-PLSR模型。如图1所示,11个主成分为最佳主成分,使用11个最佳的主成分建立PLSR模型即为O-PLER模型。
参见图2,图2为100次模型的预测均方根误差分布图,从上述MCCV的具体步骤可以看出,异常样本的识别或者异常样本的最终确定都取决于模型的预测精度,所以所建模型不稳定或者预测精度不高,容易造成对异常样本的误判或多判。因此在利用O-PLSR模型进行异常点判定之前,需验证O-PLSR模型的稳定性。为此,利用随机法从106个样本中随机选取80个样本作为校正集,其余26个样本作为预测集,重复100次试验。100次模型的预测均方根误差分布图如图2所示,从图中可以看出,100次模型的SEP值较均匀的分布在0.2到0.65之间,标准差仅为0.087,说明O-PLSR所建模型的预测准确且稳定,不会随着校正集样本的变化发生较大的变化,不会影响后续异常样本的判定。
参见图3和图4,首先用随机法按3:1的比例随机划分校正集与预测集并建立O-PLSR模型。重复此方法建模1000次;然后计算每个模型的SEP值;最后将1000个模型按照SEP值从小到大的顺序排序,并计算样本的累计频率。图3是各样本的累计频率f随排序后模型数量的增加而变化的情况,图4为前450个模型各样本的累计频率,并对累计频率低于65%和高于85%的样本进行标注。结合图3和图4可以看出,大部分样本的累计频率随模型数目的增加逐步趋于取样频率75.5%,只有11、84和96号样本的累计频率相比于其它样本存在一定差异。这三个样本在450次模型中出现的频率分别是96.67%、92.67%和99.33%。由于11、84和96号样本出现在SEP值较小的模型中的概率更大,说明它们对低SEP值的模型具有强影响。因此,将11、84和96号样本判定为强影响点。
该方法利用二审判别法判别强影响点中的异常值,具体为利用随机法划分样本集合,校正集中有83个样本,包含11、84和96号样本,预测集使用组外的10个样本。然后分别将11、84和96号样本以及它们不同的组合从校正集中剔除,建立O-PLSR模型,并分别考察模型对校正集与预测集的预测效果,结果如表1所示,当分别删除11、96号样本时,模型的预测效果均优于序号1和2对应处理的结果,而删除84号样本时,模型的预测效果相反;并且同时删除11、96号样本的建模效果均优于84号与11、96号样本随意组合被删除的建模效果。由此可以得出11和96号样本对模型产生坏的影响,84号样本对模型产生好的影响。综上所述,当将11和96号样本置于校正集中,不仅不会对模型有贡献,还会产生坏的影响,84号样本会对模型有贡献,所以判定11和96号样本为异常样本。
表1 11、84和96号样本对模型结果的影响
其中,随机删除1个的做法是从校正集中随机删除一个除强影响点以外的样本、重复200次实验取的平均结果。随机删除2个和3个同理。表中它的值也都是进行200次实验取的平均结果。
参见图5,为了考察异常样本是否剔除完全,同时检验MCCV法寻找异常样本的可靠性,做法如下:从106个样本中剔除11号和96号再次进行MCCV实验,方法同上。图5为前450个模型各样本的累计频率,并对样本累计频率低于65%和高于85%的样本进行标注。此次实验得到10、12和84号样本为强影响点,这3个样本在450次模型中样本出现的频次分别为95.56%、100%、100%(因为去除了106个样本中的11号与96号样本,所以图5中的10、11、83号样本对应于原数据中的10、12、84号样本)。
接下来对强影响点(10、12、84号)进行二审判别。如表2所示,删除10、12、84号样本均降低了模型的预测精度,所以这三个样本对模型具有好的影响,非异常样本。此次验证并未找到新的异常样本,证明利用MCCV方法找异常样本具有可靠性。
表2 10、12和84号样本对模型结果的影响
本实施例以小麦样本蛋白质化学值和光谱数据作为本发明的实例,实践检验本发明的运用效果。实例中使用组外判别法确定最佳主成分并建立PLSR模型,即O-PLSR模型,达到了对模型稳定性的要求。通过MCCV建立大量的O-PLSR模型,识别出强影响点,并结合二审判别法对强影响点进行异常判别。通过剔除异常样本再次进行MCCV实验,结果未发现新的异常样本,验证了利用MCCV方法找异常样本具有可靠性。
以上对本发明所提供的一种基于蒙特卡洛交叉验证的NIRS异常样本的检测方法,进行了详细介绍,本文中应用了具体个例对本发明的原理及实施方式进行了阐述,以上实施例的说明只是用于帮助理解本发明的方法及其核心思想;同时,对于本领域的一般技术人员,依据本发明的思想,在具体实施方式及应用范围上均会有改变之处,综上所述,本说明书内容不应理解为对本发明的限制。
- 还没有人留言评论。精彩留言会获得点赞!