基于文本挖掘的G蛋白偶联受体药物靶标分子的向量生成方法与流程
本发明涉及基于文本挖掘的向量生成方法,特别涉及基于文本挖掘的G蛋白偶联受体药物靶标分子的向量生成方法,属于机器学习的技术领域。
背景技术:
在文本挖掘领域,一般由机器学习算法来处理由自然语言构成的文本,但自然语言往往都是无结构或者半结构化形式,需要使用特定的数学模型才能被计算机识别和处理。目前,已有众多的文本表示模型供使用,其中向量空间模型是使用最广泛、效果最好的,它的可计算性和可操作性远远优于其他模型。因此,将自然语言转化为向量的形式是文本挖掘的必经之路。
G蛋白偶联受体(即GPCR)具有七跨膜结构,是细胞信号传导过程中的重要蛋白质,它在人类视觉、嗅觉、味觉以及神经传递等各项正常生理活动和疾病过程中都发挥着重要的作用。当与外部匹配的配体结合时,激活G蛋白可用于传导细胞外部的信号,从而引起细胞内的一系列变化。GPCR是靶酶中的一种,是多种药物的靶标。现代新药研究与开发的关键首先是寻找、确定和制备药物筛选靶-分子药靶。药物靶标是指药物在体内的作用结合位点,包括基因位点、受体、酶、离子通道、核酸等生物大分子。确定新的有效药靶是新药开发的首要任务。目前已发现作为治疗药物靶标的总数约500个,而GPCR靶标占绝大多数。但仍存在一部分不知道内源配体的孤儿GPCR。
基于文本的信息将GPCR表示成向量是因为现存的关于GPCR的文本信息、参考文献等数量多且信息量大,所以将其表示为向量形式,便于分析、统计学习。GPCR药物靶标分子与配体药物的结合有很大的重要性,然而许多GPCR已存的结构、序列、生物信息不足(尤其是结构信息的缺失),导致大多数GPCR并不知道它的配体。利用大量的文本信息,将GPCR转化为向量,运用机器学习的知识,可以用来寻找孤儿GPCR的内源性配体,确定新的药物靶标以便用于新药的开发,可以用来预测靶标分子的生物学功能,预测与配体药物的结合,还可以用来寻找相似的靶标分子。
传统的向量生成方式产生的向量比较稀疏且庞大,在构建语言模型时,一方面会造成维数灾难,另一方面可能会出现“词汇鸿沟”现象,即:任意两个词之间都是孤立的,例如,“话筒”和“麦克风”表示同样的语义,但是在向量空间他们之间的距离相差很大,所以这样的话,用向量的信息就反映不出语义上的相关性。目前,已被发现的人的药物靶标分子总数约500个,其中GPCR占绝大多数。然而,目前已知GPCR药物靶标分子中,很多生物学功能不清晰,三级结构未知,尤其是存在众多的孤儿GPCR分子,其内源配体不清楚。同时,我们注意到,PubMed数据库中存在大量GPCR相关文献。而本发明有效的利用了大量的GPCR相关文本信息能够很好地解决上面的问题。
技术实现要素:
本发明目的在于针对上述现有技术的不足,提出了一种基于文本挖掘的G蛋白偶联受体药物靶标分子的向量生成方法,该方法基于文本挖掘的向量生成,把对文本内容的处理简化为向量空间中的向量运算,而向量空间上的相似度可以用来表示文本语义上的相似度。该方法应用于G蛋白偶联受体GPCR,使每个GPCR用一个向量表示,能够较为全面准确的表示出每个GPCR的重要特征,以便确定新的药靶应用于新药物的开发。
本发明解决其技术问题所采取的技术方案是:本发明通过从大量的医学文献库中训练出文本的模型,使GPCR的每一个重要的特征用一个低维向量表示,每一个特征向量看成一个示例,那么每个GPCR就可以看作是一个多示例的包,运用多示例单示例化,在考虑每一个特征的前提下,将每个GPCR包中的多个特征向量转化为一个单一向量。
方法流程:
步骤1:提取G蛋白偶联受体GPCR的多个关键描述信息;
步骤2:从PubMed数据库中得到关于受体的所有文献的摘要信息;
步骤3:利用Word2vec和Doc2vec工具将GPCR的多个关键描述信息转化为多个向量;
步骤4:将GPCR的多个向量单示例化,即一个GPCR用一个向量表示。
进一步地,本发明所述方法通过从大量的医学文献库中训练出文本的模型,使GPCR的每一个重要的特征用一个低维向量表示,每一个特征向量看成一个示例,那么每个GPCR就可以看作是一个多示例的包,运用多示例单示例化,在考虑每一个特征的前提下,将每个GPCR包中的多个特征向量转化为一个Fisher单一向量。
进一步地,本发明所述方法从信息最丰富、资源最广的蛋白质数据库即uniprot_GPCR数据库中提取出每一个GPCR的蛋白质、物种、分子功能、参考文献等相关特征信息。
进一步地,本发明所述方法是以国际上公认的最具权威的生物医学文献数据库即PubMed数据库中关于受体的所有文献信息约100万篇摘要文献作为训练集。
进一步地,本发明的训练的文本是PubMed数据库中的摘要文献,目标域数据是提取出的每一个GPCR的关键信息,即蛋白质、物种、分子功能、参考文献,词语采用word2vec工具训练,句子采用Doc2vec的训练方法,将词语与句子分开训练,句子的向量不再单纯的是将句中词语向量求平均,文本的信息比较全面。
进一步地,本发明所述方法将GPCR看成一个包,它的多个关键信息向量看成多个示例,用多示例压缩技术将多示例的包数据压缩成单示例的Fisher单一向量,最终使一个GPCR用一个向量表示,得到的向量是基于语义空间得到,向量能够反映出词语语义空间的信息。
本发明还提供了一种基于文本挖掘的G蛋白偶联受体药物靶标分子的向量生成系统,该系统包括词向量模型的训练过程模块、句子向量模型的训练过程模块、多示例单示例化过程模块。
词向量模型的训练过程模块是从PubMed生物文献数据库中下载关于GPCR的摘要文献作为训练集,从uniprot_GPCR数据库中提取出的每一个GPCR的蛋白质、物种等信息作为需要求的目标向量,运用word2vec将大量的文献信息训练成词向量。
句子向量模型的训练过程模块不同于词向量的训练过程,采用另外的训练方式,即Doc2vec。以PubMed生物文献数据库中下载摘要文献作为训练集,从uniprot_GPCR数据库中提取出的每一个GPCR的分子功能、参考文献等句子信息作为需要求的目标向量。
多示例单示例化过程模块是将一个GPCR看成一个多示例的包,根据每一个GPCR的蛋白质、物种、分子功能、参考文献等多个向量信息看成多个示例,将这些多示例转化成单示例,即一个GPCR的包用一个向量表示。
有效效果:
1、本发明是基于大量的文本,运用单词转换为向量word2vec模型来提取GPCR的特征,由于训练过程中利用了词的上下文,丰富了语义信息,提高了样本特征提取的准确度。另外,本发明对于句子采用句子转化为向量Doc2vec模型训练,克服了忽略上下文和单词顺序信息的缺点,保留了句中许多重要的信息。
2、本发明将GPCR转化为向量,因为已知GPCR药物靶标分子中,很多生物学功能不清晰,三级结构未知,尤其是存在众多的未知内源配体的孤儿GPCR分子。将GPCR转化为向量,运用机器学习的知识,可以用来寻找孤儿GPCR的内源配体,确定新的药物靶标应于新药的开发,还可以用来预测靶标分子的生物学功能,预测与配体药物的结合,另外可以用来寻找相似的靶标分子。
3、本发明是基于文本空间得到的GPCR的实数值向量,因为现存的关于GPCR的文本文献、参考文献等数量多且信息量大,基于文本得到的向量准确度高,信息覆盖全面,而且文本内容的处理简化为向量空间中的向量运算,向量的距离能够体现文本语义上的相似度。
4、本发明运用多示例单示例化技术将得到描述GPCR的若干个向量转换成一个单一向量,使得每一个GPCR用一个向量表示,扩展了应用范围。
5、本发明能够有效地利用大量的GPCR相关文本信息,将基于文本生成向量的方式与多示例单示例化算法结合到一起,生成的向量能够反映GPCR药物靶标分子在功能上的相似度。
附图说明
图1为本发明系统的架构图。
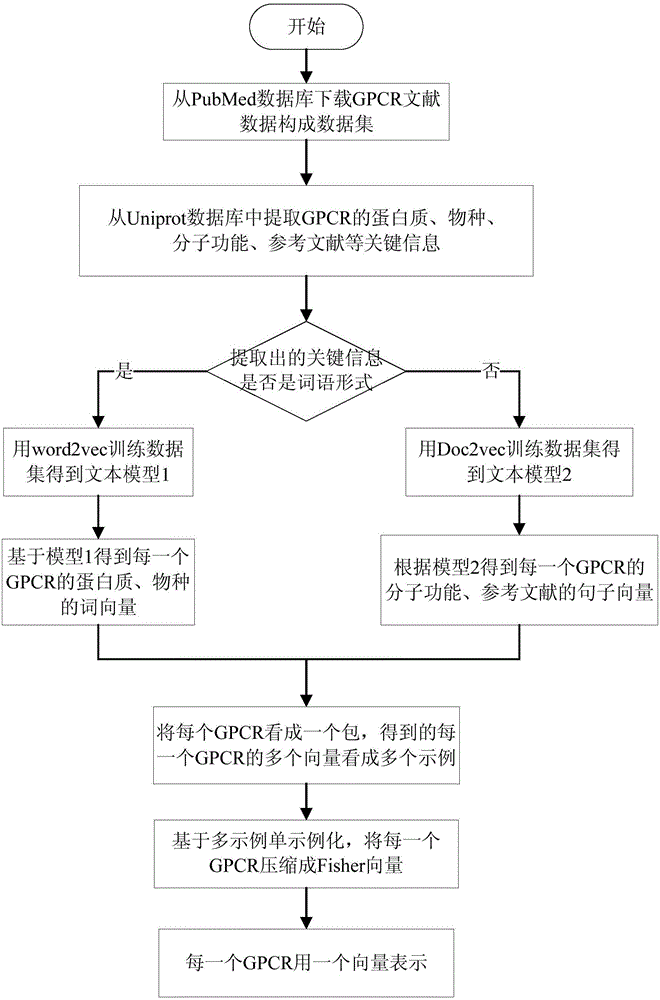
图2为本发明的方法流程图。
图3为本发明的GPCR包多示例单示例化原理说明流程图。
具体实施方式
以下结合说明书附图对本发明创造作进一步的详细说明。
实施例一
如图1所示,本发明提供了一种基于文本挖掘的G蛋白偶联受体药物靶标分子的向量生成系统,该系统包括词向量模型的训练过程模块、句子向量模型的训练过程模块、多示例单示例化过程模块。
词向量模型的训练过程模块是从PubMed生物文献数据库中下载关于GPCR的约100万篇摘要文献作为训练集,从uniprot_GPCR数据库中提取出的每一个GPCR的蛋白质、物种等信息作为需要求的目标向量,运用word2vec将大量的文献信息训练成词向量。word2vec是将单词训练成实数值向量的高效工具,它是在训练语言模型的同时,顺便得到词向量的,它可以根据上下文预测当前词,也可以根据当前词预测上下文的语境。
以下是已知上下文,估算当前词语的语言模型。其学习目标是最大化对数似然函数:
其中,C为语料库,也就是本发明中的100万篇摘要文献数据库,w表示语料库C中任意一个词,Context为词w的上下文语境。
此模型有输入层、投影层和输出层,输入层为上下文的词语的词向量(训练开始的时候,词向量是个随机值,随着训练的进行不断被更新)。投影层就是对输入的词向量相加求和。输出层对应为Huffman树,也就是除了根结点以外树中的每个结点都取0、1编码,每个叶子结点代表语料库中的一个词语,各词在语料中出现的次数作为每个结点的权值。对于词库中的词w,Huffman树中必存在一条从根结点到词w对应叶子结点的路径pw,此路径上存在bw-1个分支,即为bw-1个0、1编码,每个分支看成一次二分类,每次分类就会产生一个概率,计算目标词产生的概率就是从根结点到目标词w的结点这条路径上每次二分类概率的乘积。因此,w的条件概率可以转化为:
其中,xw为输入层向量的累加和,就是投影层的向量,bw为路径中包含结点的个数,表示路径pw中第i个结点对应的编码,表示路径pw中第i个节点的父结点对应的向量,因为根节点无编码,故编码从下标2开始,对应的参数向量下标从1开始(根节点为1)。每一项是个二分类将编码为0的结点定义为正类,编码为1的结点定义为负类,也就是:
二分类问题可以用逻辑斯谛回归表示,正类表示为σ(x),则负类为1-σ(x),因此,每个结点产生的概率可以表示为:
于是目标函数转化为:
为了最大化目标函数,采用随机梯度下降法,就是对目标函数中的相关参数xw、做一次刷新,因此需要考虑关于这些向量的梯度。
为方便求梯度,将上式中求和符号内的内容简化为L(w,i)
计算L(w,i)关于的梯度:
于是,的更新公式为:
其中α表示学习率。接下来计算L(w,i)关于xw的梯度:
不过xw是上下文的词向量的和,不是上下文单个词的词向量,因此采取的是直接将xw的更新量整个应用到每个单词的词向量上去:
其中,代表上下文中某一个单词的词向量。运用此模型能够得到提取出的G蛋白偶联受体(GPCR)的蛋白质、有机体等信息的向量。
句子向量模型的训练过程模块运用Word2vec得到词向量,提取出GPCR的分子功能、参考文献等信息为句子的形式,为了得到这些句子的向量,采用另外的训练方式即Doc2vec。仍然以PubMed生物文献数据库中下载关于GPCR的100万篇摘要文献作为训练集,每一个GPCR的分子功能、参考文献等句子信息作为需要求的目标向量。
Doc2vec是在word2vec基础上提出的另一个用于计算长文本向量的工具,它是根据上下文将一个句子或段落表征为实数值向量,它的工作原理与word2vec极为相似,也是在训练语言模型的同时,得到了句子向量,不同的是在输入层word2vec将词语的初始向量作为输入,而Doc2vec将目标词的句子初始值向量和词语初始值向量一起作为输入。和Word2Vec一样,该模型也可以在给定上下文和句子的情况下预测目标单词的概率。在一个句子或者文档的训练过程中,给每一个段落分配一个固定的id,每一个段落映射为一个唯一的向量,段落向量组成一个矩阵D,每个段落是其中的一列。
模型也分为输入层、投影层和输出层,输入层为段落矩阵D和上下文词语矩阵W,投影层就是对输入的句子向量和词向量相加求和,输出层为目标词语的向量。当给定一系列训练的词和段落w1,w2,...,wT,Dt,模型的目标是最大化平均对数似然函数:
其中,wt为所求的目标词,Dt为目标词所在的段落向量。预测的目标可以通过多类分类器即softmax函数(softmax相当于是逻辑斯谛模型在多分类问题上的推广,逻辑斯谛模型是针对二分类的,而softmax是针对多分类的)计算,因此,当已知上下文和段落后,目标词的生成概率可以表示为:
其中yi是输出的目标词为语料库中第i个词语的概率,也就是:
y=b+Uh(wt-k,...,wt+k,Dt;W,D) (12)
其中,b是softmax函数的偏差参数,U为映射层到输出层的权重,h是从词矩阵W和段落矩阵D中输入的词向量和段落向量的均值。
Doc2vec训练过程中新增了段落位置id,即训练语料库中每个句子都有一个唯一的段落id。段落和普通的词一样,也是先映射成一个向量,即段落向量。段落向量与词向量的维数虽一样,但是来自于两个不同的向量空间。在训练过程中,段落向量和词向量累加或者连接起来,作为输出层softmax的输入。在一个句子或者文档的训练过程中,段落id保持不变,相当于每次在预测单词的概率时,都利用了整个句子的语义。在预测阶段,给待预测的句子新分配一个段落id,词向量和输出层softmax的参数保持训练阶段得到的参数不变,重新利用梯度下降训练待预测的句子。待收敛后,即得到待预测句子的段落向量。
Doc2vec相比于word2vec模型,区别点为:在Doc2vec里,输入包括段落和词语,训练后的结果不仅更新了词语的向量,还能得到段落的向量。
多示例单示例化过程模块是将一个GPCR看成一个多示例的包,根据以上过程得到的每一个GPCR的蛋白质、物种、分子功能、参考文献等多个向量信息看成多个示例,将这些多示例转化成单示例,即一个GPCR的包用一个向量表示。
解决多示例转化成单示例的一个重要问题是需要充分考虑到每一个示例对包的贡献。将一个包中的多个示例压缩成单示例会造成一定的信息损失,但是使得多示例问题得以简化,提高了算法效率。本发明中采用将多示例转化为一个Fisher向量。
Fisher向量是将原始样本映射到特征空间上得到的特征向量,它能把各个大小不同的描述子集转化成大小一致的一个向量,可以应用于多示例组成的包转化为一个特征向量表达。利用Fisher向量来代替原本的包,这就完成了对多示例的压缩。对于包它的Fisher向量计算方式如下:
由于包中的示例都是独立同分布的,因此使用混合高斯模型,包Xi取自这个混合高斯模型的概率为:
其中,p是整个样本集的概率密度函数,ωk是混合权重,μk是平均向量,∑k是第k个高斯过程的协方差对角矩阵。通过在样本集上进行最大似然估计得到混合高斯模型的参数λ={ωk,μk,Σk,k=1,...,K},假设通过混合高斯模型得到了K个高斯分布,pk为第k个高斯模型的概率密度函数,则有:
ωk需要满足以下条件:
由贝叶斯定理得到第xij个示例取自第k个高斯分布的概率为:
接下来利用以上的概率密度函数计算N的Fisher向量:
Fisher向量实际上描述的是混合高斯模型该如何调整参数λ,才能使得Xi完全取自混合高斯模型分布。
本发明将每一个G蛋白偶联受体GPCR包中所有的特征向量转化为一个Fisher单一向量描述。
本发明的特征提取方面基于文本挖掘的方式,以生物医学的100万篇生物文献数据库作为训练集,运用word2vec以及Doc2vec将每一个GPCR的多个特征高效的转化为实数值向量。
本发明根据提取的多个特征,将每一个特征向量看成一个示例,那么每个GPCR就可以看作是一个多示例的包,运用多示例单示例化技术,将每个GPCR的多个特征转化为一个单一向量。
综上所述,本发明一方面是因为存的关于GPCR的文本信息量多且信息量大,另一方面,GPCR药物靶标与配体药物的结合很重要,然而,目前已知GPCR药物靶标分子中,很多生物学功能不清晰,三级结构未知,尤其是存在众多的不知道内源配体的孤儿GPCR分子。同时,我们注意到,PubMed数据库中存在大量GPCR相关文献。利用大量的文本信息,将GPCR转化为向量,运用机器学习的知识,可以用来寻找孤儿GPCR的内源配体,确定新的药物靶标分子以便用于新药的开发,还可以用来预测靶标分子的新的生物学功能,预测与配体药物的结合,还可以用来寻找相似的靶标分子等等,扩展了GPCR的应用范围。
实施例二
如图2所示,本发明提供了一种基于文本挖掘的G蛋白偶联受体药物靶标分子的向量生成方法,该方法包括如下步骤:
步骤1:从提供生物医学方面的论文搜寻以及摘要的PubMed生物文献数据库中下载关于GPCR的相关文献摘要,大约有100万篇,构建成文本模型的训练集。
步骤2:从信息最丰富、资源最广的蛋白质数据库,即uniprot数据库中提取出每一个GPCR的蛋白质、物种、分子功能、参考文献(不止一个)等相关特征信息,这些信息能够相对全面的描述每一个G蛋白偶联受体。
步骤3:根据上述步骤1中已经构建好的训练集,利用谷歌开发出的将单词转换成向量形式的Word2vec工具训练出文本的模型,建立一个文本模型文件,此文件存储了100万篇摘要中大多数词语的向量。
步骤4:将提取出的每一个GPCR的蛋白质、物种等特征信息根据步骤3训练出的文本模型转换成实数值向量,而对于描述GPCR的分子功能、参考文献等句子信息运用Doc2vec将这些句子转化成低维实数值向量。
步骤5:将每一个GPCR看成一个包,根据步骤3、步骤4得到的关于每个GPCR的多个实数值向量看成多个示例,利用多示例单示例化的方法将描述每个GPCR的若干个向量转换成一个Fisher单一向量。即一个GPCR只用一个向量表示,且全面的涵盖了描述它的大多数信息。
进一步地,本发明上述步骤4中,Doc2vec的训练集也是生物文献数据库中100万篇摘要文献,但得到的是句子或段落的向量。
如图3所示,将每一个GPCR看成一个包,一个GPCR的多个向量看成多个示例,构建GPCR的数据集D,即为D={(Xi,yi),i=1,...,N},其中Xi为第i个GPCR,yi表示其对应的标记,xij表示第i个GPCR中的第j个示例。根据GPCR的数据集构建混合高斯模型,使用最大似然估计确定混合高斯模型的参数λ,调整λ使每一个GPCR的包能完全取自混合高斯模型分布。对λ的参数求偏导归一化的值即为Fisher单一向量,也就是每一个GPCR转化为一个向量。
- 还没有人留言评论。精彩留言会获得点赞!
- 5‑HT2B拮抗剂的制造方法与工艺
- 1‑(噻唑‑2‑基)哌啶‑4‑醇的合成方法与制造工艺
- 一种大麻酚类化合物的合成方法与制造工艺
- 一类对GPR84具有激动作用的化合物及其制备方法和用途与制造工艺
- eGFP标记GPR120阳性细胞的小鼠模型构建方法
- 靶向生长抑素受体的Al<sup>18</sup>F?NOTA?PEG<sub>6</sub>?TATE及其制备方法和应用
- 吡咯烷衍生物的口服制剂的制作方法
- 胰高血糖素样肽-1受体激动剂制备治疗肺动脉高压药物的应用
- 一种armc5基因检测试剂盒及突变位点的制作方法
- 作为mglu5受体的负性别构调节剂的6-氟-2-[4-(吡啶-2-基)丁-3-炔-1-基]咪唑并[1,2a ...的制作方法