一种非小细胞肺癌驱动基因筛选与功能分析的方法与流程
本发明属于生物技术领域,涉及一种非小细胞肺癌驱动基因筛选与功能分析的方法,具体地说,涉及一种利用多种数据库进行非小细胞肺癌驱动基因筛选与功能分析的方法。
背景技术:
肺癌是中国,以及全球发病率及死亡率第一的恶性肿瘤。在过去的40年间,肺癌的5年生存率仅从12%上升至16%,最主要原因是诊断时已属晚期,相反,早期诊断的肺癌进行手术后生存率可提高到80%。可见,早发现、早期诊断对肺癌的治疗及预后具有重要的临床意义。当前广泛运用的检测手段包括无创检查(如X线、CT、钼靶摄片等)和有创检查(纤维支气管镜、支气管造影、B超或CT定位下穿刺活检等),但缺乏依从性和普及运用的可能。因此,探讨肺癌分子致病机理并找寻新的肺癌分子标志物,让肺癌患者能够及时有效的早查、早诊、早治,是提高肺癌患者生存率、降低死亡率的关键科学问题。
尽管目前有一些肿瘤标志物,如CA125(癌抗原125)、CA19-9(癌抗原19-9)、CEA(癌胚抗原)等可用于肺癌的检测,但敏感性和特异性均不高,所以目前为止,尚没有理想的可供临床使用的肺癌早期筛查和诊断标志物。不断地发现和鉴定新的肺癌相关癌基因/蛋白仍是一项重要的工作。癌症通常是由于DNA突变的积累造成的。驱动基因(Driver gene)的突变是导致癌症的最主要原因,到目前为止,研究者致力于找到导致癌症的个体基因和细胞的改变途径。近年来,临床已经发现了一系列肺癌的驱动基因,包括ALK、EGFR、BRAF、KRAS及HER2突变等。在亚裔肺腺癌患者中,87%的患者被发现已知驱动基因,其中81%的驱动基因已有明确的靶向抑制剂。因此,驱动基因的发现可以为探讨癌症发病机制、寻找个体化治疗靶点,以及筛选潜在的肿瘤诊断标志物提供理论依据。
肿瘤基因组学综合数据库(The Integrative Oncogenomics database,IntOGen)整合了用组织分类的多维的人类肿瘤基因组学数据,其中组织名符合ICD-O命名规范。IntOGen数据库使不同变异类型(如基因突变,拷贝数改变)的数据挖掘成为可能。数据库操作简单,数据全面,免费共享,并为后期数据挖掘和信息推广提供了良好的平台。IntOGen数据库在分子生物学领域中有着广泛的应用前景,为癌症驱动基因的挖掘与筛选提供了最佳平台。
非小细胞肺癌(Non-Small Cell Lung Cancer,LSCLC)是肺癌的主要病理类型,本发明通过对IntOGen数据库中NSCLC(包括肺腺癌(Lung Adenocarcinoma,LUAD)和肺鳞癌(Lung Squamous Cell Carcinoma,LUSC))的基因突变数据进行挖掘,寻找肺鳞癌和肺腺癌共有的驱动基因,并利用DAVID和STRING数据库对这些基因进行功能富集和通路分析。本发明旨在提供一种利用多种数据库挖掘癌症驱动基因的方法,为非小细胞肺癌分子机理的探讨与临床肿瘤标志物的筛选提供可行的方法和依据。
技术实现要素:
本发明的目的在于提供一种非小细胞肺癌驱动基因筛选与功能分析的方法,利用IntOGen数据库分别筛选肺鳞癌和肺腺癌驱动基因,通过Venn图制作确定肺鳞癌和肺腺癌共有的驱动基因,利用DAVID数据库对筛选出的驱动基因进行GO功能富集分析和KEGG通路富集分析,利用STRING数据库进行蛋白间相互作用分析,为NSCLC的肿瘤标志物筛选、分子发病机制等提供有意义的探索和依据。
其具体技术方案为:
一种非小细胞肺癌驱动基因筛选与功能分析的方法,包括以下步骤:
1)利用IntOGen数据库筛选NSCLC两个亚型肺鳞癌和肺腺癌的驱动基因:在IntOGen数据库中查找“lung adenocarcinoma”和“lung squamous cell carcinoma”,获得肺腺癌和肺鳞癌驱动基因的信息,肺腺癌纳入了两个研究平台391个样本的数据,肺鳞癌纳入了一个研究平台174个样本的数据。共发现181个肺腺癌驱动基因和147个肺鳞癌驱动基因。
2)利用Venn图制作确定肺鳞癌和肺腺癌共有的驱动基因:为了寻找NSCLC,即肺腺癌和肺鳞癌,共有的驱动基因,利用Venn图在线制作工具寻找两个集合中共有的基因。结果发现91个肺鳞癌和肺腺癌共有的驱动基因。其中突变率比较高的有TP53,CDKN2A,KEAP1,NF1,RB1等。
3)利用生物信息学技术进行GO基因富集功能和KEGG通路分析:利用DAVID在线软件对91个驱动基因进行生物信息学分析,为NSCLC标志物筛选及分子机制研究提供依据。基因本体论数据库可以对基因和蛋白功能进行描述和限定,GO包括了三级结构的标准语言,主要包括分子功能(molecular function,MF)、生物学途径(biological process,BP)和细胞学组件(cell component,CC)。通过DAVID在线软件选择GO数据库,生物学过程分析表明这些驱动基因共参与了64种分子功能,主要涉及protein binding(蛋白结合),poly(A)RNA binding(poly(A)RNA结合),ATP binding(ATP结合),protein kinase binding(蛋白激酶结合),chromatin binding(染色质结合),receptor signaling protein serine/threonine kinase activity(受体信号蛋白丝氨酸/苏氨酸激酶活性),protein phosphatase binding(蛋白磷酸酶结合),identical protein binding(相同蛋白结合),transcription factor binding(转录因子结合),cadherin binding involved in cell-cell adhesion(钙粘蛋白结合参与的细胞间粘附)等。参与了51种生物学途径,主要涉及positive regulation of transcription from RNA polymerase II promoter(RNA聚合酶II启动子的转录正调节),positive regulation of transcription(转录正调节),embryonic cranial skeleton morphogenesis(胚胎颅骨骨骼形态发生),Ras protein signal transduction(Ras蛋白信号转导),in utero embryonic development(子宫内胚胎发育),viral process(病毒过程),MAPK cascade(MAKP级联)等。参与了34种细胞组件的构成,主要包括nucleoplasm(细胞核浆质),cytosol(胞质溶胶),nucleus(细胞核),cytoplasm(细胞浆),cell-cell adherens junction(细胞粘附连接),focal adhesion(焦点粘附),membrane(膜)等。KEGG通路分析结果发现,这些基因主要参与Pathways in cancer(癌症通路),Pancreatic cancer(胰腺癌),Prostate cancer(前列腺癌),Melanoma(黑色素瘤),Non-small cell lung cancer(非小细胞肺癌),MAPK signaling pathway(MAKP信号通路)等重要通路,其中EGFR,CDKN2A,KRAS,MAP2K1,TP53,RB1,PIK3R3,STK4基因参与了NSCLC通路。
4)驱动基因的蛋白互相作用网络图绘制
当蛋白质相互作用网络被破坏时,会引发细胞功能的障碍。研究这些相互作用有助于构建相关的网络模型,从而对细胞甚至疾病发生的分子机制进行解释。因此,蛋白质相互作用网络在生物学研究中具有极为重要的作用。
STRING(http://string-db.org/)数据库是一个目前常用的搜寻已知蛋白质和预测蛋白质相互作用软件系统。该相互作用包括了蛋白质之间直接的相互作用,也包括蛋白质间接功能的相关性。将91个驱动基因上传至STRING在线工具,分析这些基因编码的蛋白质间的相互作用,整个网络以TP53、KRAS、EGFR、PTEN、CTNNB1、RB1、SMAD4、HSP90AA1、HSP90AB1、MET等蛋白为核心,与其他10个以上的蛋白存在相互作用关系,而这些蛋白正是癌症通路和NSCLC通路的主要蛋白,这些基因在NSCLC的发病机制中发挥着重要的作用,有可能成为NSCLC的潜在诊断标志物和治疗靶点。
与现有技术相比,本发明的有益效果为:
本发明挖掘并筛选NSCLC的驱动基因,并进行生物信息学分析。希望能从对NSCLC的生物学性质,以及NSCLC发生、发展过程中基本的分子机制的研究得到深刻认识,为NSCLC的诊断提供检测标志物及新的治疗点,也为疾病的预防和治疗等提供可靠的科学依据。
附图说明
图1为非小细胞肺癌驱动基因筛选及功能分析流程图;
图2为利用IntOGen进行肺腺癌分析结果;
图3为肺腺癌LUAD_TCGA研究的详细信息;
图4为肺腺癌LUAD_WUST研究的详细信息;
图5为肺鳞癌LUSC_TCGA研究详细信息;
图6为IntOGen数据库获得肺腺癌驱动基因云图;
图7为IntOGen数据库获得肺鳞癌驱动基因云图;
图8为Venn图分析肺腺癌和肺鳞癌共有的驱动基因;
图9为91个驱动基因编码的蛋白质间相互作用(PPI)结构图。
具体实施方式
下面结合附图和具体实施例对本发明的技术方案作进一步详细地说明。
实施例1.
一种利用多种数据库筛选非小细胞肺癌(NSCLC)驱动基因并进行功能富集分析和通路分析的方法(图1),包括以下步骤:
1.利用IntOGen数据库筛选NSCLC驱动基因:
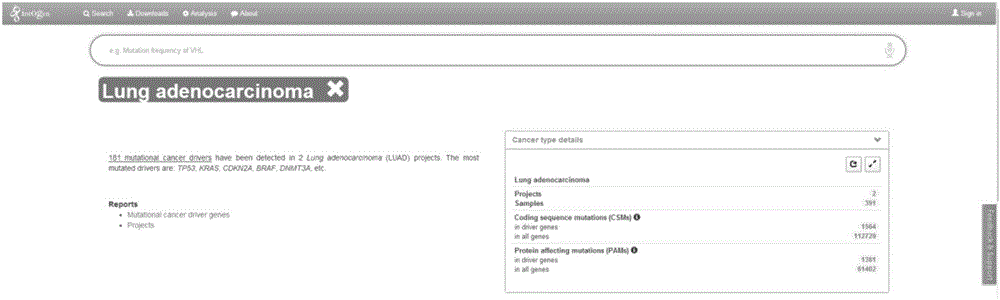
1)利用IntOGen数据库筛选NSCLC两个亚型(肺鳞癌和肺腺癌)的驱动基因:IntOGen数据库是一个用于鉴定不同肿瘤驱动基因的Web平台,它汇集了目前可利用的癌症基因突变的大数据库,并可在线进行系统分析。在IntOGen数据库(http://www.intogen.org/mutations/)中查找“lung adenocarcinoma”和“lung squamous cell carcinoma”,获得肺腺癌和肺鳞癌驱动基因的信息,肺腺癌纳入了两个研究平台391个样本的数据(LUAD_TCGA和LUAD_WUSTL),共发现181个肺腺癌驱动基因(图2)。其中,肺腺癌LUAD_TCGA研究数据来自TCGA数据库,检测了228个样本,在22638个基因中发现了169个肺腺癌驱动基因(图3);肺腺癌LUAD_WUSTL研究数据来自Nature杂志,是威斯康星大学基因组中心的研究结果,在547个基因中发现了23个肺腺癌驱动基因(图4)。肺鳞癌纳入了一个研究,LUSC_TCGA研究来自TCGA数据库,检测了174个样本,在20703个基因中发现了147个肺鳞癌驱动基因(图5)。
IntOGen数据库可以绘制驱动基因云图,通过基因名称字体的大小体现基因突变频率的高低,图6为181个肺腺癌驱动基因云图,图7为147个肺鳞癌驱动基因云图。另外,在IntOGen数据库中还可以下载各个驱动基因的突变频率,结果显示,肺腺癌突变率比较高的驱动基因有TP53,KRAS,STK11,EGFR和MLL等(表1),肺鳞癌突变率比较高的驱动基因为TP53,MLL,PIK3CA,CDKN2A和NFE2L2等(表2)。
表1.IntOGen筛选的肺腺癌突变率前20位的驱动基因
C:clustered mutations(聚类突变);F:Functional mutations(功能突变);R:Recurrent mutations(回复突变)
表2.IntOGen筛选的肺鳞癌率突变率前20位的驱动基因
C:clustered mutations(聚类突变);F:Functional mutations(功能突变);R:Recurrent mutations(回复突变)
2)利用Venn图制作确定肺鳞癌和肺腺癌共有的驱动基因:Venn图(Venn diagram)是在所谓的集合论(或者类的理论)数学分支中,用以表示集合(或类)的一种草图。它们用于展示在不同的事物群组(集合)之间的数学或逻辑联系。为了寻找NSCLC,即肺腺癌和肺鳞癌,共有的驱动基因,本发明利用Venn图在线制作工具(http://bioinformatics.psb.ugent.be/webtools/Venn/)寻找两个集合中共有的基因。结果发现91个肺鳞癌和肺腺癌共有的驱动基因(图8)。其中突变率比较高的有TP53,CDKN2A,KEAP1,NF1,RB1等。
3.利用DAVID数据库进行基因富集功能分析:
应用生物信息学方法分析生物数据,提出与疾病发生、发展相关的基因或基因集,再进行实验验证,是一条高效的研究途径。本发明以IntOGen数据库中关于NSCLC的驱动基因为分析材料,利用Venn图在线分析工具筛选出NSCLC驱动基因,再利用DAVID在线分析网络平台对差异表达基因进行生物信息学分析,为NSCLC标志物筛选及分子机制研究提供依据。
DAVID生物信息数据库(the Database for Annotation,Visualization and Integrated Discovery),是一个基于web的一种基因功能富集分析软件,整合了生物学数据库功能注释和信息链接为特点覆盖广泛的分析工具,使用者只需要提供一份基因列表,便可以应用提供的分析内容和分析工具,实现各项功能注释分析和整合,从统计学层面关联到最显著富集的生物学注释。分析的结果可以与其他的数据库链接。使用在线分析软件对选出的目的基因的KEGG通路、本体论的细胞成分、分子功能、生物过程进行分类、定义和注释。
1)GO功能注释:基因本体论(Gene Ontology,简称GO)数据库室友基因本体论联合会所建立,该数据库可以对基因和蛋白功能进行描述和限定,GO包括了三级结构的标准语言,主要包括如下:
分子功能(molecular function,MF):它包括基因产物的功能,如与碳水化合物结合或ATP水解酶活性等;生物学途径(biological process,BP):它是分子功能的组合,可获得更广的生物功能,如嘿岭代谢或分子代谢。细胞学组件(cell component,CC):包括了亚细胞结构、位置和大分子复合物,如高尔基体、端粒和识别起始的复合物等。
本发明中获得的是一组基因,对它们进行直接的功能注释,得到的功能节点数量庞大,且互相交叠,该将导致分析结果冗余。因此,我们选择对数据进行功能富集分析。该方法可有效增加研究的可靠性,并对生物现象中相关的生物学过程作出有效识别,更有利于获得有意义的功能信息。本发明选择应用较为广泛的DAVID在线软件对91个NSCLC的驱动基因进行了GO功能富集分析。
通过DAVID生物学过程分析表明这些驱动基因共参与了64种分子功能,主要涉及protein binding(蛋白结合),poly(A)RNA binding(poly(A)RNA结合),ATP binding(ATP结合),protein kinase binding(蛋白激酶结合),chromatin binding(染色质结合),receptor signaling protein serine/threonine kinase activity(受体信号蛋白丝氨酸/苏氨酸激酶活性),protein phosphatase binding(蛋白磷酸酶结合),identical protein binding(相同蛋白结合),transcription factor binding(转录因子结合),cadherin binding involved in cell-cell adhesion(钙粘蛋白结合参与的细胞间粘附)等(表3)。参与了51种生物学途径,主要涉及positive regulation of transcription from RNA polymerase II promoter(RNA聚合酶II启动子的转录正调节),positive regulation of transcription(转录正调节),embryonic cranial skeleton morphogenesis(胚胎颅骨骨骼形态发生),Ras protein signal transduction(Ras蛋白信号转导),in utero embryonic development(子宫内胚胎发育),viral process(病毒过程),MAPK cascade(MAKP级联)等(表3)。参与了34种细胞组件的构成,主要包括nucleoplasm(细胞核浆质),cytosol(胞质溶胶),nucleus(细胞核),cytoplasm(细胞浆),cell-cell adheres junction(细胞粘附连接),focal adhesion(焦点粘附),membrane(膜)等(表3)。
表3.非小细胞肺癌驱动基因显著富集的GO功能前十位
2)KEGG通路分析
KEGG(Kyoto Encyclopedia of Genes and Genomes,京都基因与基因组百科全书)是东京大学和R本京都大学共同研制的数据库。KEGG提供查询基因代谢途径、产物、酶等作用,也通过BLAST比对查询未知序列的代谢途径信息。KEGG包括有九个相互独立的数据库,主要涵盖了三大类内容:基因信息数据库、系统信息数据库和化学信息数据库。其中,系统信息数据库指的是通路数据库、疾病数据库、模块数据库、代谢通路及同源基因数据库共四个数据库;基因信息数据库包括基因数据库、同源数据库、序列相似性数据库等九个数据库;化学信息数据库包括化合物数据库、多糖数据库、酶数据库、药物数据库、反应数据库等六个数据库。本发明中,应用DAVID在线软件选择KEGG通路数据库,探索NSCLC驱动基因的生物学通路。KEGG通路分析结果发现,这些基因主要参与Pathways in cancer(癌症通路),Pancreatic cancer(胰腺癌),Prostate cancer(前列腺癌),Melanoma(黑色素瘤),Non-small cell lung cancer(非小细胞肺癌),MAPK signaling pathway(MAKP信号通路)等重要通路,其中EGFR,CDKN2A,KRAS,MAP2K1,TP53,RB1,PIK3R3,STK4基因参与了NSCLC通路(表4)。
表4.非小细胞肺癌驱动基因显著富集的KEGG通路前二十位
4.驱动基因的蛋白互相作用网络图绘制
众所周知,人体内的蛋白和其他一些小分子不是单独起作用,而是相互作用形成一种分子相互作用网络,这种相互作用网络决定了细胞乃至组织、个体的特征。当蛋白质相互作用(protein-protein interaction,PPI)网络被破坏时,可能会引发细胞功能的障碍。因此,研究这些相互作用有助于构建相关的网络模型,从而对细胞甚至疾病发生的分子机制进行解释。蛋白质相互作用网络的研究还可以潜在的推动药物发现的实际应用,因为基于蛋白质相互作用网络的药物发现可能明确的调整了疾病相关途径,改革药物发现的途径,并优于单个靶蛋白的简单的抑制或激活功能此外,应用蛋白质相互作用网络,可以预测蛋白质功能、检测蛋白质复合物、发现未知细胞系统、构建代谢或调控途径等。因此,蛋白质相互作用网络在生物学研究中具有极为重要的作用。
STRING(http://string-db.org/)数据库是一个目前常用的搜寻已知蛋白质和预测蛋白质相互作用软件系统。该相互作用包括了蛋白质之间直接的相互作用,也包括蛋白质间接功能的相关性。它的结果来自实验数据、文本挖掘、数据库以及生物信息学预测的数据。研究染色体临近、基因融合、系统进化谱和基于芯片数据的基因共表达。系统中特用的评分机制对上述不同方法得来的结果给予一定的权重,最终给出一个综合的得分。用户不仅可以输入蛋白质名称,而且可以输入基因或氨基酸序列,用以査询相关蛋白质的相互作用的信息。通过点击软件中的“Analysis”,将会构建出相关蛋白质的相互作用网络,节点间的连线采用不同的颜色,用以表明不同的计算方法。将91个驱动基因上传至STRING在线工具,分析这些基因编码的蛋白质间的相互作用,结构如图9所示,整个网络以TP53、KRAS、EGFR、PTEN、CTNNB1、RB1、SMAD4、HSP90AA1、HSP90AB1、MET等蛋白为核心,与其他10个以上的蛋白存在相互作用关系,而这些蛋白正是癌症通路和NSCLC通路的主要蛋白,这些基因在NSCLC的发病机制中发挥着重要的作用,有可能成为NSCLC的潜在诊断标志物和治疗靶点。
以上所述,仅为本发明较佳的具体实施方式,本发明的保护范围不限于此,任何熟悉本技术领域的技术人员在本发明披露的技术范围内,可显而易见地得到的技术方案的简单变化或等效替换均落入本发明的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!