人类基因突变的HGVS名称生成及分析系统的实现方法与流程
本发明属于生物信息领域,涉及基因测序后对测序数据的分析,是一种计算基因突变的hgvs名称的技术。
背景技术:
:基因变异是在1949年被人类首次发现的。根据变异是否致病,区分了突变(mutation)和多态性位点(polymorphism)。突变一般是指特定dna序列与参考序列相比发生的致病性的改变。多态性位点即snp,指非致病的序列改变,一般将人群中发生频率大于1%的序列改变界定为snp。随着基因变异的累积,人们逐渐认识到确认变异的致病性并不是一件容易的事。condit、marshall和cotton[1-3]等研究单词“mutation”和“polymorphism”的起源、演化和含义。由于“mutation”和“polymorphism”并没有严格而清晰的界限,或者其致病性未能立刻获得确认,且在人群中发生的频率不易精确获取,人类基因组变异协会建议使用更加中性的词来表示序列的改变,如序列变异(sequencevariantorvariation)、改变(alteration)和等位基因变异(allelicvariant)等。鉴于此,在下文中,使用突变或基因变异,具有相同的含义,都不代表致病与否或致病性的程度。二十世纪60年代由victormckusick等人收集人类基因变异数据[4],并创立在线人类孟德尔遗传(onlinemendelianinheritanceinman,omim)。以后出现了人类基因突变数据库(humangenemutationdatabase,hgmd);特定基因的变异数据库,包括pahdb(pah基因数据库)、pdgene(帕金森疾病相关基因)和dgv(结构变异数据库)等。在初始阶段,这些数据库缺乏对基因变异命名的统一格式,造成了提交和使用的混乱[5]。1994年richardcotton召集遗传学家成立了人类基因组变异协会(humangeomevariationsociety,hgvs),提出关于基因变异命名方面的建议,逐渐成为遗传学领域的国际准则。根据hgvs的规则,变异的定位与参考序列相关。不同类型的参考序列有不同的标记,“c.”用于冠名编码dna序列,“g.”用于冠名基因组序列,"p."用于冠名蛋白质序列,“m.”用于冠名线粒体序列,“r.”则用于冠名rna序列。人类基因组变异协会推荐使用编码dna序列作为标准参考序列,以编码序列的起始密码子atg的a作为第一个碱基,依次排列。位于编码区上游的5’端序列以-1、-2、-3等表示,位于编码区下游的3’端序列以*1、*2、*3等表示。内含子从每个内含子序列的中间划分,前半部分以前面外显子最后一个减基位置和“+”表示,如c.79+lg>t;后半部分以后面外显子的第一个碱基位置和“-”表示,如c.80-2a>c。">"表示替换,数字表示碱基改变的范围,“del”、“dup”、“ins”和“inv”分别表示缺失、重复、插入、倒位。更详细的描述见http://www.hgvs.org/mutnomen/。hgvs的命名规则也是2015年美国医学遗传学与基因组学学会(acmg)和美国分子病理学会(amp)所发布的基因变异解读标准和指南中所推荐的命名规则[6]。该指南并指定了检测基因变异命名是否符合hgvs规则的在线工具(https://mutalyzer.nl)。对于测序所得序列,经过与参考序列比对,可在序列上发现不同于参考序列的改变,即为基因变异。从变异位点给出hgvs名称,根据hgvs名称从数据库中检索变异,给出相关信息,这是基因测序分析人员的主要任务。根据参考序列及人工获取hgvs命名是可行的,但比较费时且容易出错;而使用https://mutalyzer.nl网站的“descriptionextractor”工具进行hgvs名称自动生成,又存在不能区分内含子和外显子的重大缺陷。因此我们开发了人类基因突变分析系统,用于计算基因变异的hgvs名称,并有据此调出hgmd数据库结果的能力,为基因检测的科研工作者和基因检测临床分析人员提供更多选择。参考文献:[1]condit,cm.,p.j.achter,i.lauer,ande.sefcovic,thechangingmeaningsof"mutation:"acontextualizedstudyofpublicdiscourse.hummutat,2002.19(1):69-75.[2]marshall,j.h.,onthechangingmeaningsof"mutation".hummutat,2002.19(1):76-78.[3]cotton,r.g.,communicating"mutation:"modemmeaningsandconnotations.hummutat,2002.19(1):2-3.[4]ring,h.z.,p.y.kwok,andr.g.cotton,humanvariomeproject:aninternationalcollaborationtocataloguehumangeneticvariation.pharmacogenomics,2006.7(7):969-972.[5]丛培宽,全基因组外显子测序发现x连锁显性遗传性高度近视疾病的致病基因及人类基因变异数据库lovd的创建,中国知网,硕博论文,2014[6]richardss,azizn,bales,bickd,dass,gastier-fosterj,grodyww,hegdem,lyone,spectore10,voelkerdingk,rehmhl;acmglaboratoryqualityassurancecommittee.standardsandguidelinesfortheinterpretationofsequencevariants:ajointconsensusrecommendationoftheamericancollegeofmedicalgeneticsandgenomicsandtheassociationformolecularpathology.genetmed.2015may;17(5):405-24.doi:10.1038/gim.2015.30.epub2015mar5.技术实现要素:为了诊断遗传病、癌症等,需要对目的基因测序,将测序所得序列与参考序列比对后,对于所发现的变异,用户希望尽快地和尽可能准确地获取该变异的hgvs名称、致病性信息、遗传方式信息等数据。因此我们设计了人类基因突变分析系统。人类基因变异分析系统典型的实现方案是由数据输入界面、基因信息(含序列)数据库、基因遗传方式数据库、hgmd数据库、hgvs计算程序及数据输出界面构成。在非典型的实现方案中,对这些数据库的增删,顺序调整,数据来源调整等,不构成对本
发明内容的根本改变。在数据输入界面,系统规定的典型数据格式是:基因名称,(变异位点参考序列/变异位点突变序列)变异位点之后一段序列,变异位点是纯合型还是杂合型(用“c”代表纯合型,用“z”代表杂合型)。这些信息可简化为:“gene(r/m)sz/c”。在此格式中,gene是不可缺失的,且应使用标准的基因名称。r为变异位点参考序列,m为变异位点突变序列,二者以斜杠隔开且必须同时存在。r和m皆可以代表一个碱基,也可以代表多个碱基,r和m缺失时可以用“-”代替。例如碱基缺失时,m为“-”,碱基插入时,r为“-”。z/c代表碱基杂合型或纯合型,通过峰图即可简单判断,只允许输入单个字母,也可以不输入。典型的数据输入格式之外的其它的非典型数据输入格式,包括对数据项目的增删、顺序调整、格式调整等,不构成对本
发明内容的根本改变。对于所输入的信息,人类基因突变分析系统中的hgvs运算程序根据括号中的斜杠两边的碱基数量、是否存在短杠(“-”),以及括号后提供的序列在基因组序列中的情况来判断突变型,根据括号后序列在基因组序列中的位置及突变类型来判断突变在cds坐标上的位置,从而计算出hgvs名称。在我们的典型设计方案中,本系统所能分析的基因突变类型包括:碱基置换(substitutions)、缺失(deletion)、重复(duplication)、插入(insertion)、倒位(inversion)、缺失/插入(indels)。在非典型的实现方案中,对以上突变类型数目的增删,不构成对本
发明内容的根本改变。在算出hgvs名称后,hgvs运算程序可据此从数据库中调出该突变的相关信息。在典型的实现方案中,所输出的信息包括:突变基因,转录本,核苷酸变化,氨基酸变化,纯合/杂合,遗传方式,hgmd致病性,hgmd疾病,文献等,并将这些信息输出到系统的输出界面。在非典型的实现方案中,对这些数据项目的增删、顺序调整、格式调整等,不构成对本
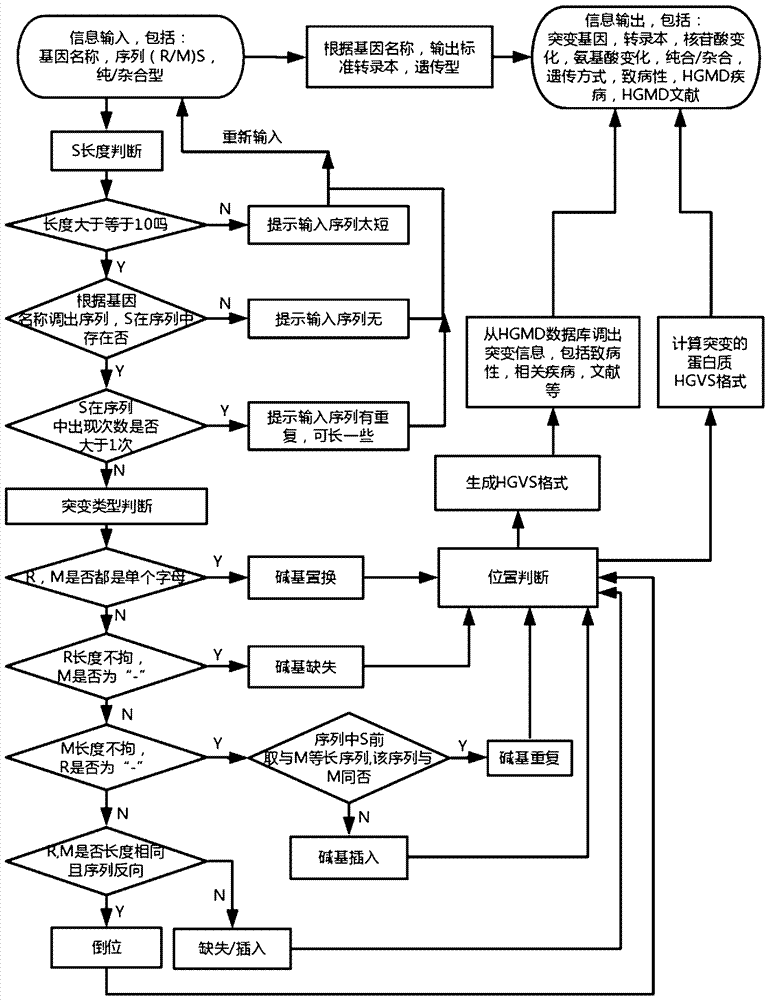
发明内容的根本改变。附图说明图1,人类基因hgvs名称生成及分析系统突变分析系统的系统构架图。分别由数据输入界面、基因信息(含序列)数据库、基因遗传方式数据库、hgmd数据库、hgvs计算程序及数据输出界面构成。图2,本发明的hgvs运算程序的流程图。信息按照一定的格式从左上角输入后,经过一系列的判断,计算,数据库调取,在右上角的信息输出处得到该变异的一系列信息。具体实施方式以下具体实施的过程,采用的是以上“
发明内容”中的典型方案。应该理解,具体实施过程采用了典型方案的思路,不表示典型方案是达成本发明目的的最佳方案。应该理解,本发明即便本发明的典型方案也并不限定于本文所描述的特定的方法、设计和流程。本文使用的术语仅为描述特定实施方式,并不意在限制本发明的范围。此外,如本领域技术人员所理解的,可以采用各种不同的方式达成类似目的。例如改变编程使用的计算机语言、在不同的计算机系统中编程、在不同的机器上使用等。采用不同方式达到同样目的,不构成对本
发明内容的改变。尽管参照具体实施方式公开了本发明,但可能对所述实施方式作出各种修饰、替换和改变而不脱离随附说明书和权利要求书中描述的本发明的完整范围。基于详述、附图、实施例和权利要求,可以明白所公开的主题的其他特征、目的和优势。可利用与本文所述那些基本上类似或等同的方法来实施或测试本发明公开的主题。人类基因突变分析系统已用多种计算机语言,在多种计算机系统中实现。其中在excel环境中,通过编程实现对编码区分析的算法已经申请软件著作权。软件名称为:“人类基因编码区突变的功能分析软件”,登记号为:2015sr028115,授权日期为2015年2月9日。1.人类基因突变分析系统的设计目的人类基因突变分析系统是为一线的遗传病基因检测人员,特别是基于sanger测序进行基因检测的实验室人员设计和使用的。系统设计的首要原则是用户体验的便利性,计算结果的准确性和导出数据的权威性。在进行sanger测序时,用户通常是针对明确的基因设计引物,扩增后进行测序,并将测序文件通过比对软件与该基因的标准序列进行比对。在比对的过程中,根据测序峰图的情况,用户可甄别哪些部位是测序过程中产生的系统性错误,哪些部位是真实的变异位点。对于所发现的区别于参考序列的真实的变异位点,用户已经知道的信息包括:基因名称,变异位点参考序列,变异位点突变序列,变异位点上游和下游序列,以及通过变异位置是单峰还是套峰判断变异为杂合型还是纯合型等。其中在判断纯合型或杂合型时,由于男性x染色体上基因,显示为单峰的变异与常染色体基因的纯合型变异表现相同但归为半合型,用户无需查阅基因定位于常染色体或性染色体,只将所见的纯合型或杂合型输入,系统可自动判断,并给出是否为半合型的提示。在系统上,用户可以简单地输入:基因名称,(变异位点原始序列/变异位点突变序列)变异位点之后一段序列,变异位点是纯合型还是杂合型(用“c”代表纯合型,用“z”代表杂合型),中间以空格隔离。例如输入:ugt1a1(g/a)gagcattttacaccttgaac,希望计算后可得到:突变基因:ugt1a1转录本:nm_000463核苷酸变化:c.211g>a氨基酸变化:p.gly71arg纯合/杂合:纯合型遗传方式:arhgmd致病性:dfphgmd疾病:hyperbilirubinaemia,associationwith文献:teng,clingenet,2007希望得到的信息中,突变基因是用户输入的基因名称,转录本是用于计算hgvs名称所用的基因转录本,核苷酸变化是计算得到的基于编码序列的hgvs名称,氨基酸变化是计算得到的基于蛋白质序列的hgvs名称,纯合/杂合是用户输入的信息并经过简单判断,遗传方式是这个基因或疾病的遗传方式,hgmd致病性和hgmd疾病是判断该突变是否致病,导致什么病的信息,文献则是与这个判断相关的文献。一般来说,这些信息回答了用户所关心的关于突变的最核心问题:突变的标准hgvs名称是什么,它是致病的吗,如果致病,致病强度如何,可导致什么疾病,有什么文献支持这个判断等。hgvs网站给出了多种突变类型。其中基因微小突变包括:碱基置换(substitutions);缺失(deletion);重复(duplication);插入(insertion);倒位(inversion);缺失/插入(indels)等。本系统可对编码dna水平的这些变异方式进行分析,发生变异的位置包括编码蛋白质的外显子区域,不编码蛋白质的外显子区域,内含子区域,5’-utr区,3’-utr区等,并给出标准hgvs名称。hgvs网站还包括其它类型的变异如序列重复数量变异,易位,嵌合体,未知序列长片段变异等复杂情况,目前本系统尚不能处理。总而言之,本系统的目的,是在用户进行序列比对遇到变异时,只要发生变异的基因名称是已知的,变异的类型不特殊,变异的位置位于基因序列之内,都可以以最简单方法的方式输入系统,系统反馈用户关于这个突变的最主要的信息,实现基因突变分析的智能化、自动化。2系统架构为了实现以上目的,人类基因变异分析系统分别由数据输入界面、基因信息(含序列)数据库、基因遗传方式数据库、hgmd数据库、hgvs计算程序及数据输出界面构成。该系统的框架结构图见说明书附图1。在数据输入界面,用户需输入:基因名称,(变异位点参考序列/变异位点突变序列)变异位点之后一段序列,变异位点是纯合型还是杂合型(用“c”代表纯合型,用“z”代表杂合型)。数据输入界面支持输入多行数据的分析。基因信息数据库所含基因列表来自hgmd数据库2015年版本包含的5969个基因。基因信息包括基因名称,来自ensembl数据库的基因组序列,基因在hgmd数据库所用的转录本名称及序列等。基因的遗传方式相对复杂一些。同一基因在不同的遗传病中可以有不同的遗传方式,同一基因同一遗传病在不同的个体中也可以有不同的遗传方式。对人类的数万个基因而言,明确其遗传方式的基因约有4000个。我们用以标注基因遗传方式的信息来自omim数据库。hgmd数据库见http://www.hgmd.cf.ac.uk/ac/index.php。hgmd分为公共版(publicversion)和专业版(professionalversion)。截止到2017年4月5日,公共版和专业版分别收录了5528和7791个基因,分别包含141635和197952个突变。所收录的突变类型包括外显子错义突变,启动子区突变,剪接位点突变,插入突变,缺失突变及复杂性重组等。hgvs计算程序可将输入的信息转化为hgvs名称。由于hgmd数据库基本上遵循了hgvs的突变命名规则,因此hgvs计算程序可在生成的hgvs名称后,通过该名称从hgmd数据库中调取与该突变有关的信息。这些信息包括:突变的致病性,突变导致的疾病,有关该突变的参考文献等。hgvs计算程序计算后,将计算结果输出到输出界面,包括:突变基因,转录本,核苷酸变化,氨基酸变化,纯合/杂合,遗传方式,hgmd致病性,hgmd疾病,文献等。3hgvs名称运算流程在数据输入环节,如前所述,用户应输入:基因名称,(变异位点参考序列/变异位点突变序列)变异位点之后一段序列,纯合型/杂合型。这些信息可简化为:”gene(r/m)sz/c”。在此格式中,gene是不可缺失的,且应使用标准的基因名称。r为变异位点参考序列,m为变异位点突变序列,r和m皆可以代表一个碱基,也可以代表多个碱基,r和m在碱基缺失时可以用“-”代替。例如碱基缺失时,m为“-”,碱基插入时,r为“-”。z/c代表碱基杂合型或纯合型,通过峰图即可简单判断,应输入单个字母,也可以不输入。对于所输入的信息,hgvs运算程序根据括号中的斜杠两边的碱基数量、是否存在短杠(“-”),以及括号后提供的序列在基因组序列中的情况来判断突变型,根据括号后序列在基因组序列中的位置及突变类型来判断突变在cds坐标上的位置,从而计算出hgvs名称。其具体的运算流程见说明书附图2。在实际运算中,程序涉及的内容与判断要多于流程图。例如,如果输入的基因不在基因库中,应如何处理;如果输入的数据格式不对,应如何判断和处理;纯/杂合型未输入应如何应对;由于这些不是运算的重点,此处不再赘述。4与hgmd数据的对接通过hgvs计算程序得到某基因某变异的hgvs名称后,可从遵循了hgvs命名规则的变异数据库中检索该变异,找到后从数据库中调取与该变异有关的信息。常用的人类基因变异数据库有clinvar,hgmd等,都基本遵循了hgvs突变命名规则。此处以hgmd为例。hgmd数据库分为公共版和专业版。在公共版,对于碱基置换型的突变,数据库提供了每个突变的登记号(accessionnumber),密码子变化(codonchange),氨基酸变化(aminoacidchange),密码子位置(codonnumber),基因组坐标及hgvs的命名(genomiccoordinates&hgvsnomenclature),表型(phenotype),参考文献(reference),评论(comments)等信息。其中基因组坐标及hgvs的命名信息被掩盖了,密码子变化和氨基酸变化的信息进行了图片化处理,无法以文字格式拷贝。因此,使用hgmd公共版是不利于hgvs名称的计算机检索的。hgmd的专业版数据库除了以上信息外,还包括突变的致病性分类(variantclass)等信息。所提供的hgvs名称可拷贝。因此,通过hgvs计算测序所得hgvs名称,可对接hgmd专业版数据,获取相关信息。5人类基因突变hgvs命名系统的使用方法对于从数据输入界面输入的基因突变数据,hgvs计算程序计算hgvs名称首先依赖于所输入数据的格式。如前所述,系统规定的数据输入格式为:gene(r/m)sz/cgene为基因的标准名称,r为突变参考序列,m为突变序列,s为突变处后面的一段序列,该序列需为ncbi上gdna上的一段序列,不拘内含子还是外显子,也不拘5'utr区域或3'utr区域,但需大于10个碱基。如使用cds序列,如所圈选序列包含不同外显子,在gdna序列中不能检索,则无法计算。z/c代表突变为杂合型还是纯合型,可以不填。这种表示方式是比对时突变记录的最简单和最直便的格式。不同突变类型的输入格式简述如下:1)碱基置换情况,r和m分别是单个碱基,如atp7b(t/c)cccccagaccttctctgtgctg,表示cccccagaccttctctgtgctg序列前发生了t到c的变异。t为参考序列,c为突变序列。2)碱基缺失,缺失时m用-表示,如:atp7b(t/-)cccccagaccttctctgtgctg表示cccccagaccttctctgtgctg前面的t缺失。atp7b(cc/-)cccagaccttctctgtgctg表示cccagaccttctctgtgctg前面的两个cc缺失。应该注意的是,按照hgvs的规则,当有连续相同的多个碱基缺失一个或几个时,应按缺失后面的碱基来算。所以,agaccttctctgtgctg前面5个c缺失两个c时,应记做:atp7b(cc/-)agaccttctctgtgctg,而不是(cc/-)cccagaccttctctgtgctg否则系统会给出错误的结果。3)碱基插入,r用-表示,即序列中在无碱基处出现新的碱基,如atp7b(-/tt)agaccttctctgtgctg表示agaccttctctgtgctg前面原来没有tt,突变后多了tt。4)碱基重复,输入格式与碱基插入相同。如插入的序列正好与前面等长的序列相同,则系统自动判断为重复。其输入格式与插入相同。例如:atp7b(-/a)gaccttctctgtgctg所插入的a前面也是a,故判定为碱基重复。5)缺失/插入,r和m其中至少一个的碱基数目要大于1,且不构成倒位。如atp7b(ag/ttt)accttctctgtgctg表示原来accttctctgtgctg前面的ag变为ttt6)倒位,r和m的长度都必须大于1,且等长,且序列为反向,其格式与缺失/插入相同。如atp7b(ag/ga)accttctctgtgctg,系统自动判断ga是ag的倒位。应该注意的是,目前hgmd数据库并未遵循hgvs关于碱基倒位的规则。hgmd将倒位视为缺失/插入。以上数据整理如下:atp7b(t/c)cccccagaccttctctgtgctgatp7b(t/-)cccccagaccttctctgtgctgatp7b(-/tt)agaccttctctgtgctgatp7b(-/a)gaccttctctgtgctgatp7b(ag/ttt)accttctctgtgctgatp7b(ag/ga)accttctctgtgctg将这些数据输入系统,可得到以下结果。其中核苷酸变化就是编码dna的hgvs名称:突变基因转录本核苷酸变化氨基酸变化纯合/杂合遗传方式致病性hgmd疾病hgmd文献atp7bnm_000053c.3419t>cp.val1140ala需输入z或cardpwilsondiseaseliu,wjg,2004atp7bnm_000053c.3419delt无数据需输入z或cardmwilsondiseasekumar,clingenet,2005atp7bnm_000053c.3424_3425instt无数据需输入z或car无无无atp7bnm_000053c.3425dupa无数据需输入z或car无无无atp7bnm_000053c.3425_3426delaginsttt无数据需输入z或car无无无atp7bnm_000053c.3425_3426invag无数据需输入z或car无无无(如在以上输入的序列后面加上空格和c或z字母,在纯和/杂合一栏将给出纯合型或杂合型的结果)。6与“mutalyzer”结果的比较美国医学遗传学与基因组学学会(acmg)所推荐的网站https://mutalyzer.nl(以下称为m系统)是从突变序列生成hgvs名称的专用工具。使用m系统生成基于cds序列的突变的hgvs名称的方法是:打开https://mutalyzer.nl网站,点击“descriptionextractor”,将参考序列输入参考序列(referencesequence)框,再输入样品序列(samplesequence),两条序列都必须是同一基因完整的cds序列,而后点击“extractvariantdescription”,即可生成突变位点的hgvd命名格式。针对同一批的突变位点,我们用人类突变分析系统(以下称h系统)和m系统分别进行了处理,对两个系统的使用方式和所得结果进行了比较。我们选择一个比较短的名为apoc2基因的cds区,其序列为:atgggcacacgactcctcccagctctgtttcttgtcctcctggtattgggatttgaggtccaggggacccaacagccccagcaagatgagatgcctagcccgaccttcctcacccaggtgaaggaatctctctccagttactgggagtcagcaaagacagccgcccagaacctgtacgagaagacatacctgcccgctgtagatgagaaactcagggacttgtacagcaaaagcacagcagccatgagcacttacacaggcatttttactgaccaagttctttctgtgctgaagggagaggagtaa以上用于举例的apoc2基因的cds区序列,第1-55碱基属于第一外显子,第56-215碱基属于第二外显子,第216-306碱基到属于第三外显子。在使用m系统时,该序列需要始终存在于referencesequence框中。(1)碱基置换将第二个位置的t改为c,然后将改变后的序列输入样品序列(samplesequence)框中,则在m系统中,点击“extractvariantdescription”,生成的hgvs名称为:2t>c在h系统中,无需输入参考序列,只要输入“apoc2(t/c)gggcacacgac”,即可得到:c.2t>c,两者一致,而h系统更符合hgvs的规范。这是由于m系统不区分所输入的序列是gdna还是cds或其它格式,需要由输入者判断,自行在其前加入g.或c.,以分别标记序列类型,以下相同。(2)碱基缺失将第二个碱基删除后输入m系统,得到:2del;h系统的输入方法是:“apoc2(t/-)gggcacacgac”,可得到c.2delt,两者一致,而h系统显示了缺失的碱基名称,更完整。(3)碱基插入在第二个碱基后插入c,m系统得到:2_3insc,h系统的输入方法是:“apoc2(-/c)gggcacacgactc”,得:c.2_3insc,两者一致。(4)碱基重复第三四五位置是g,其后插入一个g,应为碱基重复。m系统得到:5dup;h系统的输入方法是:“apoc2(-/g)cacacgactcctc”,得:c.5dupg,两者一致,而h系统给出了重复的碱基名称,更完整。(5)碱基倒位将第11位到14位的gact变为tcag,显然是倒位,m系统得到:11_14delinstcag,m系统认为是插入缺失,h系统的输入方法是:“apoc2(gact/tcag)cctcccagctctgtttct”,得:c.11_14invgact,显然h系统符合hgvs的规则,而m系统所得结果以严格的hgvs命名规则来看是错误的。(6)插入缺失将第三个g变为ccc,m系统得:3delinsccc;h系统的输入方法是:“apoc2(g/ccc)ggcacacgactc”,得:c.3delginsccc,两者一直,而h系统给出了缺失的碱基名称,更完整。(7)重复性序列(repeatsequence)对于重复性序列,两个系统都不能以hgvs的格式处理。例如ar基因中存在两段微卫星重复序列,第一段为22个agc重复,其标准命名应该是:c.173_175[22],如减少一个agc重复,名称变为c.173_175[21]。这种变化,在两个系统中都无法生成为标准的hgvs命名格式,而是当成del处理了。两个系统的不同之处可归结为:(1)数据的输入格式不同。m系统需要输入参考序列和突变后的整个序列,而突变后的整个序列需要用户根据比对结果,在参考序列上编辑。h系统不需要输入参考序列,仅将基因名称和突变及突变后的一段序列输入即可。所输入的信息可以全部来自比对界面,而不需要另行打开基因序列的数据库。因此,从使用效果看,h系统的数据输入更为简便。(2)对内含子计算能力不同。对m系统所输入的参考序列,可以是基因组序列,也可以是cds序列。前者计算突变时给出突变的基因组名称,应以g.开头,后者计算突变时给出突变的cds名称,应以c.开头。但m系统不能区分所输入的序列是外显子还是内含子,对于内含子突变不能给出其突变的cds名称。而内含子的突变,特别是位于外显子/内含子交界处附近内含子的突变,通常对转录本的剪切方式有重要影响。这属于m系统hgvs名称计算的重大缺陷。而h系统对输入的数据,不需要用户区分序列是外显子和内含子,系统可根据输入序列在基因上的位置自动区分属于内含子还是外显子,并给出相应的hgvs名称。(3)对倒位的计算能力不同。碱基倒位是一种特殊的插入缺失,在hgvs命名规则中,该突变类型归于单独的一类。然而m系统对此仍按缺失/插入处理,这是错误的,至少是没有严格遵守hgvs的规则。而h系统可以计算倒位型的突变。(4)输出格式不同。m系统对于输入的待检测序列,可以编辑一个突变,也可以编辑多个突变,经计算后给出每个突变的hgvs名称。除此之外并不能给出突变的其它信息。而h系统可以一次输入一行待测突变,也可以输入多行突变,且多行突变可分属不同的基因。所输出的数据,除了突变在dna水平的hgvs名称外,还可以包括突变在蛋白质水平的hgvs名称,以及自动调出与该突变及该基因有关的信息。结论是:在一般意义上,人类基因突变分析系统(h系统)要优于mutalyzer系统(m系统)。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1