非水电解质二次电池的制作方法
本发明涉及非水电解质二次电池。
背景技术:
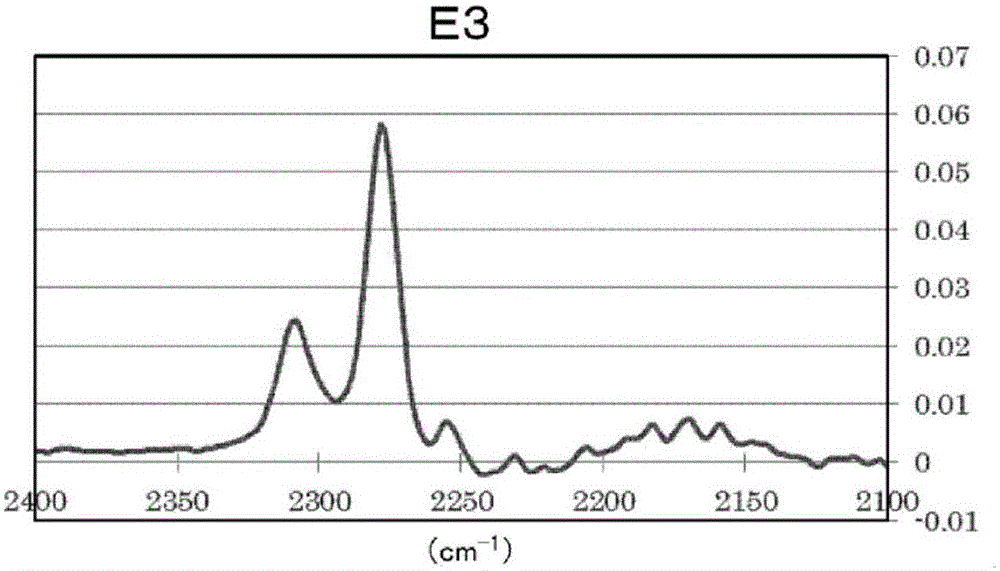
:已知在非水电解质二次电池的负极和正极的表面生成被膜。该被膜也被称为SEI(SolidElectrolyteInterphase:固体电解质界面膜),由电解液的还原分解物等构成(例如,参照专利文献1)。以下,根据情况,将该被膜简称为SEI被膜。负极表面和正极表面的SEI被膜允许锂离子等电荷载体的通过。另外,认为例如负极表面的SEI被膜存在于负极表面与电解液之间,有助于抑制电解液的进一步还原分解。特别是对于使用了石墨、Si系的负极活性物质的低电位负极而言,SEI被膜是必须的。认为如果由于SEI被膜存在而抑制电解液的继续分解,则能够提高经过循环后的电池的放电特性(以下,称为循环特性)。然而其另一方面,对于现有的非水电解质二次电池而言,并不能说负极表面和正极表面的SEI被膜一定有助于电池特性的提高。因此,希望开发出一种具有能够使电池特性进一步提高的SEI被膜的非水电解质二次电池。另一方面,例如锂离子二次电池是充放电容量高、能够高输出功率化的二次电池。锂离子二次电池现在主要用作便携式电子设备、笔记本电脑、电动汽车用的电源,需要更小型·轻型的二次电池。特别是在汽车用途,由于需要以大电流进行锂离子二次电池的充放电,所以要求开发出一种具有高的输入输出特性的锂离子二次电池。锂离子二次电池的正极和负极分别具有能够吸留和放出锂(Li)的活性物质。而且,是通过锂离子在密封于两极间的电解液内移动而工作的。为了提高输入输出特性等锂离子二次电池的电池特性,需要改进正极和/或负极中使用的活性物质、粘结剂以及改进电解液等。作为锂离子二次电池的负极活性物质,为了避免树枝状晶体析出的问题,广泛使用石墨等碳材料。为了使锂离子可逆地对这样的负极活性物质插入和脱离,一般的电解液中使用环状酯、链状酯等非水系的碳酸酯系溶剂。然而对于使用碳酸酯系溶剂的现有的电解液而言,难以大幅提高作为锂离子二次电池的输入输出特性的一种的倍率特性。即,如下述的非专利文献1~3所述,使用了碳酸亚乙酯、碳酸亚丙酯等碳酸酯系溶剂的锂离子二次电池中反应电阻大。因此,为了改进倍率容量特性,需要从根本上重新考虑电解液的溶剂组成。现有技术文献专利文献专利文献1:日本特开2007-19027号公报专利文献2:日本特开2007-115671号公报专利文献3:日本特开2003-268053号公报专利文献4:日本特开2006-513554号公报非专利文献非专利文献1:T.Abeetal.,J.Electrochem.Soc.,151,A1120-A1123(2004).非专利文献2:T.Abeetal.,J.Electrochem.Soc.,152,A2151-A2154(2005).非专利文献3:Y.Yamadaetal.,Langmuir,25,12766-12770(2009).技术实现要素:本发明是考虑了上述情况而进行的,要解决的课题是得到电池特性优异的非水电解质二次电池。已知在非水电解质二次电池的负极和正极的表面生成被膜。该被膜也被称为SEI(SolidElectrolyteInterphase),由电解液的还原分解物等构成。例如,日本特开2007-19027号公报中也介绍了该被膜。以下,根据情况,将该被膜简称为SEI被膜。负极表面和正极表面的SEI被膜允许锂离子等电荷载体的通过。另外,认为例如负极表面的SEI被膜存在于负极表面与电解液之间,有助于抑制电解液的进一步还原分解。特别是对于使用了石墨、Si系的负极活性物质的低电位负极而言,SEI被膜是必须的。认为如果由于SEI被膜存在而抑制电解液的继续分解,则能够提高经过循环后的电池的放电特性(以下称为循环特性)。然而其另一方面,对于现有的非水电解质二次电池而言,并不能说负极表面和正极表面的SEI被膜一定有助于电池特性的提高。因此,希望开发出一种具有能够使电池特性进一步提高的SEI被膜的非水电解质二次电池。本发明的发明人等经过深入研究,结果发现在现有的非水电解质二次电池中,根据SEI被膜的组成、结构、厚度有时锂离子等电荷载体的通过性不充分,SEI被膜会成为非水电解质二次电池的反应电阻增大(例如输入输出特性的降低)的原因。这样,以开发出具有能够抑制电解液的继续分解且电荷载体的透过性也优异的SEI被膜的非水电解质二次电池为目标,作了进一步的研究。其结果,发现在使用特殊的电解液的非水电解质二次电池中,在负极表面生成来自该电解液的特殊结构的SEI被膜。另外,进一步发现在正极表面也生成来自该电解液的特殊结构的SEI被膜。而且发现具有该电解液和来自该电解液的特殊结构的SEI被膜的非水电解质二次电池的寿命、输入输出特性等电池特性优异。即,解决上述课题的本发明的非水电解质二次电池(1)包括正极、电解液和负极,上述电解液含有盐和具有杂元素的有机溶剂,该盐以碱金属、碱土金属或铝为阳离子且在阴离子的化学结构中含有硫元素和氧元素,对于上述电解液的振动光谱中的来自上述有机溶剂的峰强度,将上述有机溶剂本来的峰的强度设为Io、将上述峰位移后的峰的强度设为Is时,满足Is>Io,在上述负极的表面形成了具有S=O结构的含S、O被膜。另外,解决上述课题的本发明的非水电解质二次电池(1)包括正极、电解液和负极,上述电解液含有盐和具有杂元素的有机溶剂,该盐以碱金属、碱土金属或铝为阳离子且在阴离子的化学结构中含有硫元素和氧元素,对于上述电解液的振动光谱中的来自上述有机溶剂的峰强度,将上述有机溶剂本来的峰的强度设为Io、将上述峰产生位移后的峰的强度设为Is时,满足Is>Io,在上述负极的表面和上述正极的表面中的至少上述正极的表面形成了具有S=O结构的含S、O被膜。这样的非水电解质二次电池(1)在负极表面和/或正极表面具有特殊结构的SEI被膜,即含S、O被膜,电池特性优异。另一方面,一般的负极是通过在集电体涂布含有负极活性物质和粘结剂的浆料并干燥而制作的。粘结剂具有使负极活性物质之间粘结和使活性物质与集电体之间粘结的作用、覆盖保护负极活性物质的作用。作为以往使用的负极用粘结剂,有聚偏氟乙烯(PVdF)等含氟系聚合物、羧甲基纤维素(CMC)等水溶性纤维素衍生物、聚丙烯酸等。例如在上述的专利文献2中,记载了含有选自聚丙烯酸和聚甲基丙烯酸中的聚合物且该聚合物含有酸酐基的锂离子二次电池用负极。另外,在上述的专利文献3中,记载了将丙烯酸与甲基丙烯酸共聚而得的聚合物用作负极用粘结剂或正极用粘结剂。此外,在上述的专利文献4中,记载了将丙烯酰胺和丙烯酸和衣康酸共聚而得的聚合物用作负极用粘结剂或正极用粘结剂。解决上述课题的本发明的非水电解质二次电池(2)的特征具备电解液和负极,上述电解液含有以碱金属、碱土金属或铝为阳离子的盐和具有杂元素的有机溶剂,对于振动光谱中的来自上述有机溶剂的峰强度,将上述有机溶剂本来的峰的强度设为Io、将上述峰位移后的峰的强度设为Is时,满足Is>Io,上述负极具备含有粘结剂的负极活性物质层,该粘结剂由具有亲水基团的聚合物构成。本发明的非水电解质二次电池(2)使用具有亲水基团的聚合物作为负极用的粘结剂,并且使用本发明的电解液作为电解液。使用聚偏氟乙烯等聚合物作为负极用的粘结剂时,即便使用相同的本发明的电解液,也难以兼具倍率特性的提高和循环特性的提高。但是,通过使用由具有亲水基团的聚合物构成的粘结剂作为负极用粘结剂,能够兼具倍率特性的提高和循环特性的提高。作为其理由,如下考虑:例如如果非水电解质二次电池为锂离子二次电池,则由于粘结剂所含的羧基等极性基团吸引锂离子,所以在浓度过电压主导的高倍率侧负荷特性提高。另外认为粘结剂产生的活性物质保护作用使循环特性提高。也就是说,如果采用这样的非水电解质二次电池(2),则利用电解液和粘结剂的最佳组合能够实现倍率容量特性的提高,并且还能够改进循环特性。以下,根据需要,有时将“含有以碱金属、碱土金属或铝为阳离子的盐和具有杂元素的有机溶剂,且对于振动光谱中的来自上述有机溶剂的峰强度,将上述有机溶剂本来的峰的强度设为Io、将上述峰位移后的峰的强度设为Is时,满足Is>Io的电解液”称为“本发明的电解液”。此外,有时将在上述的本发明的电解液中盐的阴离子的化学结构中含有硫元素和氧元素的电解液特别称为“电解液(1)”或“本发明的电解液(1)”。本发明的电解液(1)是本发明的电解液的一种,在上述非水电解质二次电池(1)中含有。当然,非水电解质二次电池(2)也可以含有本发明的电解液(1)。此外,根据需要,将非水电解质二次电池(1)和非水电解质二次电池(2)统称为本发明的非水电解质二次电池。本发明的非水电解质二次电池的电池特性优异。附图说明图1是电解液E3的IR光谱。图2是电解液E4的IR光谱。图3是电解液E7的IR光谱。图4是电解液E8的IR光谱。图5是电解液E10的IR光谱。图6是电解液C2的IR光谱。图7是电解液C4的IR光谱。图8是乙腈的IR光谱。图9是(CF3SO2)2NLi的IR光谱。图10是(FSO2)2NLi的IR光谱。图11是电解液E11的IR光谱。图12是电解液E12的IR光谱。图13是电解液E13的IR光谱。图14是电解液E14的IR光谱。图15是电解液E15的IR光谱。图16是电解液C6的IR光谱。图17是碳酸二甲酯的IR光谱。图18是电解液E16的IR光谱。图19是电解液E17的IR光谱。图20是电解液E18的IR光谱。图21是电解液C7的IR光谱。图22是碳酸甲乙酯的IR光谱。图23是电解液E19的IR光谱。图24是电解液E20的IR光谱。图25是电解液E21的IR光谱。图26是电解液C8的IR光谱。图27是碳酸二乙酯的IR光谱。图28是(FSO2)2NLi的IR光谱(1900~1600cm-1)。图29是电解液E8的拉曼光谱。图30是电解液E9的拉曼光谱。图31是电解液C4的拉曼光谱。图32是电解液E11的拉曼光谱。图33是电解液E13的拉曼光谱。图34是电解液E15的拉曼光谱。图35是电解液C6的拉曼光谱。图36是评价例8的对反复快速充放电的响应性的结果。图37是评价例12中的针对实施例1-1、实施例1-2和比较例1-1的负极含S、O被膜的碳元素的XPS分析结果。图38是评价例12中的针对实施例1-1、实施例1-2和比较例1-1的负极含S、O被膜的氟元素的XPS分析结果。图39是评价例12中的针对实施例1-1、实施例1-2和比较例1-1的负极含S、O被膜的氮元素的XPS分析结果。图40是评价例12中的针对实施例1-1、实施例1-2和比较例1-1的负极含S、O被膜的氧元素的XPS分析结果。图41是评价例12中的针对实施例1-1、实施例1-2和比较例1-1的负极含S、O被膜的硫元素的XPS分析结果。图42是评价例12中的实施例1-1的负极含S、O被膜的XPS分析结果。图43是评价例12中的实施例1-2的负极含S、O被膜的XPS分析结果。图44是评价例12中的实施例1-1的负极含S、O被膜的BF-STEM图像。图45是评价例12中的针对实施例1-1的负极含S、O被膜的C的STEM分析结果。图46是评价例12中的针对实施例1-1的负极含S、O被膜的O的STEM分析结果。图47是评价例12中的针对实施例1-1的负极含S、O被膜的S的STEM分析结果。图48是评价例12中的针对实施例1-1的正极含S、O被膜的O的XPS分析结果。图49是评价例12中的针对实施例1-1的正极含S、O被膜的S的XPS分析结果。图50是评价例12中的针对实施例1-4的正极含S、O被膜的S的XPS分析结果。图51是评价例12中的针对实施例1-4的正极含S、O被膜的O的XPS分析结果。图52是评价例12中的针对实施例1-4、实施例1-5和比较例1-2的正极含S、O被膜的S的XPS分析结果。图53是评价例12中的针对实施例1-6、实施例1-7和比较例1-3的正极含S、O被膜的S的XPS分析结果。图54是评价例12中的针对实施例1-4、实施例1-5和比较例1-2的正极含S、O被膜的O的XPS分析结果。图55是评价例12中的针对实施例1-6、实施例1-7和比较例1-3的正极含S、O被膜的O的分析结果。图56是评价例12中的针对实施例1-4、实施例1-5和比较例1-2的负极含S、O被膜的S的分析结果。图57是评价例12中的针对实施例1-6、实施例1-7和比较例1-3的负极含S、O被膜的S的分析结果。图58是评价例12中的针对实施例1-4、实施例1-5和比较例1-2的负极含S、O被膜的O的分析结果。图59是评价例12中的针对实施例1-6、实施例1-7和比较例1-3的负极含S、O被膜的O的分析结果。图60是评价例13中的电池的复阻抗平面曲线。图61是评价例20中的实施例1-1的非水电解质二次电池的DSC图表。图62是评价例20中的比较例1-1的非水电解质二次电池的DSC图表。图63是表示评价例21中的EB4的电流与电极电位的关系的图。图64是表示评价例22中的针对EB4的电位(3.1~4.6V)与响应电流的关系的图。图65是表示评价例22中的针对EB4的电位(3.1~5.1V)与响应电流的关系的图。图66是表示评价例22中的针对EB5的电位(3.1~4.6V)与响应电流的关系的图。图67是表示评价例22中的针对EB5的电位(3.1~5.1V)与响应电流的关系的图。图68是表示评价例22中的针对EB6的电位(3.1~4.6V)与响应电流的关系的图。图69是表示评价例22中的针对EB6的电位(3.1~5.1V)与响应电流的关系的图。图70是表示评价例22中的针对EB7的电位(3.1~4.6V)与响应电流的关系的图。图71是表示评价例22中的针对EB7的电位(3.1~5.1V)与响应电流的关系的图。图72是表示评价例22中的针对CB4的电位(3.1~4.6V)与响应电流的关系的图。图73是表示评价例22中的针对EB5的电位(3.0~4.5V)与响应电流的关系的图。应予说明,图73是改变图66的纵轴的比例尺得到的图。图74是表示评价例22中的针对EB5的电位(3.0~5.0V)与响应电流的关系的图。应予说明,图74是改变图67的纵轴的比例尺得到的图。图75是表示评价例22中的针对EB8的电位(3.0~4.5V)与响应电流的关系的图。图76是表示评价例22中的针对EB8的电位(3.0~5.0V)与响应电流的关系的图。图77是表示评价例22中的针对CB5的电位(3.0~4.5V)与响应电流的关系的图。图78是表示评价例22中的针对CB5的电位(3.0~5.0V)与响应电流的关系的图。图79是评价例24中的实施例1-1的非水电解质二次电池的充放电后的铝箔的表面分析结果。图80是评价例24中的实施例1-2的非水电解质二次电池的充放电后的铝箔的表面分析结果。图81是EB9的充放电曲线。图82是EB10的充放电曲线。图83是EB11的充放电曲线。图84是EB12的充放电曲线。图85是CB6的充放电曲线。图86是评价例29的低温倍率特性的结果。图87是评价例29的低温倍率特性的结果。图88是表示实施例2-1、2-2和比较例2-1的非水电解质二次电池的充放电特性的图。具体实施方式以下,对用于实施本发明的方式进行说明。应予说明,只要没有特殊说明,则本说明书中记载的数值范围“a~b”是指在其范围内包含下限a和上限b。而且,还包括这些上限值和下限值以及实施例中列举的数值,通过将这些数值任意组合可构成数值范围。进而从数值范围内任意选择的数值可以为上限、下限的数值。本发明的非水电解质二次电池(1)包括负极、正极和本发明的电解液(1),在正极和/或负极的表面形成了含S、O被膜。另外,本发明的非水电解质二次电池(2)包括本发明的电解液和具备负极活性物质层的负极,该负极活性物质层含有由具有亲水基团的聚合物构成的粘结剂。如上所述,本发明的非水电解质二次电池(1)中,在正极和/或负极的表面形成了含S、O被膜,实现电池特性的提高。因此,非水电解质二次电池(1)中除电解液以外的电池构成要素,例如负极活性物质、正极活性物质、导电助剂、粘结剂、集电体和隔离件等没有特别限定。另外,如上所述,本发明的非水电解质二次电池(2)中,利用负极用粘结剂与电解液的最佳组合实现电池特性的提高。因此,非水电解质二次电池(2)中除负极用粘结剂和电解液以外的电池构成要素没有特别限定。任一情况下,在本发明的非水电解质二次电池中的负极表面和/或正极表面均形成特殊结构的SEI被膜,即含S、O被膜。另外,本发明的非水电解质二次电池中的电荷载体也没有特别限定。例如,本发明的非水电解质二次电池可以是以锂为电荷载体的非水电解质二次电池(例如,锂二次电池、锂离子二次电池),可以是以钠为电荷载体的非水电解质二次电池(例如,钠二次电池、钠离子二次电池)。如上所述,本发明的电解液含有以碱金属、碱土金属或铝为阳离子的盐和具有杂原子的有机溶剂,对于振动光谱中的来自有机溶剂的峰强度,将有机溶剂本来的峰的强度设为Io、将有机溶剂本来的峰产生波数位移后的峰的强度设为Is时,满足Is>Io。另外,其中,非水电解质二次电池(1)中使用的电解液(1)使用以碱金属、碱土金属或铝为阳离子且阴离子的化学结构中含有硫元素和氧元素的盐作为盐。也就是说,电解液(1)是本发明的电解液的一个方式。因此只要为本发明的电解液,则Io与Is的关系通常是Is>Io。与此相对,现有的电解液,Is与Io的关系是Is<Io。在这点上本发明的电解液与现有的电解液有很大不同。以下,根据需要,将本发明的电解液和/或电解液(1)所含的盐,即,“以碱金属、碱土金属或铝为阳离子的盐”和/或“以碱金属、碱土金属或铝为阳离子且在阴离子的化学结构中含有硫元素和氧元素的盐”有时称为“金属盐”、支持盐、支持电解质或简称为“盐”。应予说明,由于电解液(1)是本发明的电解液的一个方式,所以在没有特别说明、解释的情况下对“本发明的电解液”进行说明的位置就是对包含电解液(1)在内的本发明的电解液整体进行说明。〔金属盐〕本发明的电解液中的金属盐只要是通常作为电池的电解液所含的LiClO4、LiAsF6、LiPF6、LiBF4、LiAlCl4等电解质使用的化合物即可。作为金属盐的阳离子,可举出锂、钠、钾等碱金属,铍、镁、钙、锶、钡等碱土金属和铝。金属盐的阳离子优选为与使用电解液的电池的电荷载体相同的金属离子。例如,如果使用本发明的电解液作为锂离子二次电池用的电解液,则金属盐的阳离子优选为锂。此时,优选盐的阴离子的化学结构含有选自卤素、硼、氮、氧、硫或碳中的至少一种元素。具体例示含有卤素或硼的阴离子的化学结构,可举出ClO4、PF6、AsF6、SbF6、TaF6、BF4、SiF6、B(C6H5)4、B(oxalate)2、Cl、Br、I。对于含有氮、氧、硫或碳的阴离子的化学结构,以下进行具体说明。盐的阴离子的化学结构优选为下述通式(1)、通式(2)或通式(3)表示的化学结构。(R1X1)(R2X2)N······通式(1)(R1选自氢、卤素、可以被取代基取代的烷基、可以被取代基取代的环烷基、可以被取代基取代的不饱和烷基、可以被取代基取代的不饱和环烷基、可以被取代基取代的芳香族基团、可以被取代基取代的杂环基、可以被取代基取代的烷氧基、可以被取代基取代的不饱和烷氧基、可以被取代基取代的硫代烷氧基、可以被取代基取代的不饱和硫代烷氧基、CN、SCN、OCN。R2选自氢、卤素、可以被取代基取代的烷基、可以被取代基取代的环烷基、可以被取代基取代的不饱和烷基、可以被取代基取代的不饱和环烷基、可以被取代基取代的芳香族基团、可以被取代基取代的杂环基、可以被取代基取代的烷氧基、可以被取代基取代的不饱和烷氧基、可以被取代基取代的硫代烷氧基、可以被取代基取代的不饱和硫代烷氧基、CN、SCN、OCN。另外,R1和R2可以相互键合而形成环。X1选自SO2、C=O、C=S、RaP=O、RbP=S、S=O、Si=O。X2选自SO2、C=O、C=S、RcP=O、RdP=S、S=O、Si=O。Ra、Rb、Rc、Rd各自独立地选自氢、卤素、可以被取代基取代的烷基、可以被取代基取代的环烷基、可以被取代基取代的不饱和烷基、可以被取代基取代的不饱和环烷基、可以被取代基取代的芳香族基团、可以被取代基取代的杂环基、可以被取代基取代的烷氧基、可以被取代基取代的不饱和烷氧基、可以被取代基取代的硫代烷氧基、可以被取代基取代的不饱和硫代烷氧基、OH、SH、CN、SCN、OCN。另外,Ra、Rb、Rc、Rd可以与R1或R2键合而形成环。)R3X3Y······通式(2)(R3选自氢、卤素、可以被取代基取代的烷基、可以被取代基取代的环烷基、可以被取代基取代的不饱和烷基、可以被取代基取代的不饱和环烷基、可以被取代基取代的芳香族基团、可以被取代基取代的杂环基、可以被取代基取代的烷氧基、可以被取代基取代的不饱和烷氧基、可以被取代基取代的硫代烷氧基、可以被取代基取代的不饱和硫代烷氧基、CN、SCN、OCN。X3选自SO2、C=O、C=S、ReP=O、RfP=S、S=O、Si=O。Re、Rf各自独立地选自氢、卤素、可以被取代基取代的烷基、可以被取代基取代的环烷基、可以被取代基取代的不饱和烷基、可以被取代基取代的不饱和环烷基、可以被取代基取代的芳香族基团、可以被取代基取代的杂环基、可以被取代基取代的烷氧基、可以被取代基取代的不饱和烷氧基、可以被取代基取代的硫代烷氧基、可以被取代基取代的不饱和硫代烷氧基、OH、SH、CN、SCN、OCN。另外,Re、Rf可以与R3键合而形成环。Y选自O、S。)(R4X4)(R5X5)(R6X6)C······通式(3)(R4选自氢、卤素、可以被取代基取代的烷基、可以被取代基取代的环烷基、可以被取代基取代的不饱和烷基、可以被取代基取代的不饱和环烷基、可以被取代基取代的芳香族基团、可以被取代基取代的杂环基、可以被取代基取代的烷氧基、可以被取代基取代的不饱和烷氧基、可以被取代基取代的硫代烷氧基、可以被取代基取代的不饱和硫代烷氧基、CN、SCN、OCN。R5选自氢、卤素、可以被取代基取代的烷基、可以被取代基取代的环烷基、可以被取代基取代的不饱和烷基、可以被取代基取代的不饱和环烷基、可以被取代基取代的芳香族基团、可以被取代基取代的杂环基、可以被取代基取代的烷氧基、可以被取代基取代的不饱和烷氧基、可以被取代基取代的硫代烷氧基、可以被取代基取代的不饱和硫代烷氧基、CN、SCN、OCN。R6选自氢、卤素、可以被取代基取代的烷基、可以被取代基取代的环烷基、可以被取代基取代的不饱和烷基、可以被取代基取代的不饱和环烷基、可以被取代基取代的芳香族基团、可以被取代基取代的杂环基、可以被取代基取代的烷氧基、可以被取代基取代的不饱和烷氧基、可以被取代基取代的硫代烷氧基、可以被取代基取代的不饱和硫代烷氧基、CN、SCN、OCN。另外,R4、R5、R6中任二个或三个可以键合而形成环。X4选自SO2、C=O、C=S、RgP=O、RhP=S、S=O、Si=O。X5选自SO2、C=O、C=S、RiP=O、RjP=S、S=O、Si=O。X6选自SO2、C=O、C=S、RkP=O、RlP=S、S=O、Si=O。Rg、Rh、Ri、Rj、Rk、Rl各自独立地选自氢、卤素、可以被取代基取代的烷基、可以被取代基取代的环烷基、可以被取代基取代的不饱和烷基、可以被取代基取代的不饱和环烷基、可以被取代基取代的芳香族基团、可以被取代基取代的杂环基、可以被取代基取代的烷氧基、可以被取代基取代的不饱和烷氧基、可以被取代基取代的硫代烷氧基、可以被取代基取代的不饱和硫代烷氧基、OH、SH、CN、SCN、OCN。另外,Rg、Rh、Ri、Rj、Rk、Rl可以与R4、R5或R6键合而形成环。)对上述通式(1)~(3)表示的化学结构中的“可以被取代基取代的”这句话进行说明。例如如果是“可以被取代基取代的烷基”,则是指烷基的一个或多个氢被取代基取代的烷基或没有特别的取代基的烷基。作为“可以被取代基取代的”这句话中的取代基,可举出烷基、烯基、炔基、环烷基、不饱和环烷基、芳香族基团、杂环基、卤素、OH、SH、CN、SCN、OCN、硝基、烷氧基、不饱和烷氧基、氨基、烷基氨基、二烷基氨基、芳氧基、酰基、烷氧基羰基、酰氧基、芳氧基羰基、酰氧基、酰基氨基、烷氧基羰基氨基、芳氧基羰基氨基、磺酰基氨基、氨磺酰基、氨基甲酰基、烷基硫基、芳基硫基、磺酰基、亚磺酰基、脲基、磷酸酰胺基、磺基、羧基、异羟肟酸基、亚磺基、肼基、亚氨基、甲硅烷基等。这些取代基可以进一步被取代。另外取代基为2个以上时,取代基可以相同也可以不同。盐的阴离子的化学结构更优选下述通式(4)、通式(5)或通式(6)表示的化学结构。(R7X7)(R8X8)N······通式(4)(R7、R8各自独立地为CnHaFbClcBrdIe(CN)f(SCN)g(OCN)h。n、a、b、c、d、e、f、g、h各自独立地为0以上的整数,满足2n+1=a+b+c+d+e+f+g+h。另外,R7和R8可以相互键合而形成环,此时,满足2n=a+b+c+d+e+f+g+h。X7选自SO2、C=O、C=S、RmP=O、RnP=S、S=O、Si=O。X8选自SO2、C=O、C=S、RoP=O、RpP=S、S=O、Si=O。Rm、Rn、Ro、Rp各自独立地选自氢、卤素、可以被取代基取代的烷基、可以被取代基取代的环烷基、可以被取代基取代的不饱和烷基、可以被取代基取代的不饱和环烷基、可以被取代基取代的芳香族基团、可以被取代基取代的杂环基、可以被取代基取代的烷氧基、可以被取代基取代的不饱和烷氧基、可以被取代基取代的硫代烷氧基、可以被取代基取代的不饱和硫代烷氧基、OH、SH、CN、SCN、OCN。另外,Rm、Rn、Ro、Rp可以与R7或R8键合而形成环。)R9X9Y······通式(5)(R9为CnHaFbClcBrdIe(CN)f(SCN)g(OCN)h。n、a、b、c、d、e、f、g、h各自独立地为0以上的整数,满足2n+1=a+b+c+d+e+f+g+h。X9选自SO2、C=O、C=S、RqP=O、RrP=S、S=O、Si=O。Rq、Rr各自独立地选自氢、卤素、可以被取代基取代的烷基、可以被取代基取代的环烷基、可以被取代基取代的不饱和烷基、可以被取代基取代的不饱和环烷基、可以被取代基取代的芳香族基团、可以被取代基取代的杂环基、可以被取代基取代的烷氧基、可以被取代基取代的不饱和烷氧基、可以被取代基取代的硫代烷氧基、可以被取代基取代的不饱和硫代烷氧基、OH、SH、CN、SCN、OCN。另外,Rq、Rr可以与R9键合而形成环。Y选自O、S。)(R10X10)(R11X11)(R12X12)C······通式(6)(R10、R11、R12各自独立地为CnHaFbClcBrdIe(CN)f(SCN)g(OCN)h。n、a、b、c、d、e、f、g、h各自独立地为0以上的整数,满足2n+1=a+b+c+d+e+f+g+h。R10、R11、R12中任二个可以键合而形成环,此时,形成环的基团满足2n=a+b+c+d+e+f+g+h。另外,R10、R11、R12三个可以键合而形成环,此时,三个中二个基团满足2n=a+b+c+d+e+f+g+h,一个基团满足2n-1=a+b+c+d+e+f+g+h。X10选自SO2、C=O、C=S、RsP=O、RtP=S、S=O、Si=O。X11选自SO2、C=O、C=S、RuP=O、RvP=S、S=O、Si=O。X12选自SO2、C=O、C=S、RwP=O、RxP=S、S=O、Si=O。Rs、Rt、Ru、Rv、Rw、Rx各自独立地选自氢、卤素、可以被取代基取代的烷基、可以被取代基取代的环烷基、可以被取代基取代的不饱和烷基、可以被取代基取代的不饱和环烷基、可以被取代基取代的芳香族基团、可以被取代基取代的杂环基、可以被取代基取代的烷氧基、可以被取代基取代的不饱和烷氧基、可以被取代基取代的硫代烷氧基、可以被取代基取代的不饱和硫代烷氧基、OH、SH、CN、SCN、OCN。另外,Rs、Rt、Ru、Rv、Rw、Rx可以与R10、R11或R12键合而形成环。)上述通式(4)~(6)表示的化学结构中的“可以被取代基取代的”这句话的意思与在上述通式(1)~(3)中说明的意思相同。上述通式(4)~(6)表示的化学结构中,n优选为0~6的整数,更优选为0~4的整数,特别优选为0~2的整数。应予说明,上述通式(4)~(6)表示的化学结构的R7和R8键合或R10、R11、R12键合而形成环时,n优选为1~8的整数,更优选为1~7的整数,特别优选为1~3的整数。盐的阴离子的化学结构进一步优选下述通式(7)、通式(8)或通式(9)表示的化学结构。(R13SO2)(R14SO2)N······通式(7)(R13、R14各自独立地为CnHaFbClcBrdIe。n、a、b、c、d、e各自独立地为0以上的整数,满足2n+1=a+b+c+d+e。另外,R13和R14可以相互键合而形成环,此时,满足2n=a+b+c+d+e。)R15SO3·····通式(8)(R15为CnHaFbClcBrdIe。n、a、b、c、d、e各自独立地为0以上的整数,满足2n+1=a+b+c+d+e。)(R16SO2)(R17SO2)(R18SO2)C·····通式(9)(R16、R17、R18各自独立地为CnHaFbClcBrdIe。n、a、b、c、d、e各自独立地为0以上的整数,满足2n+1=a+b+c+d+e。R16、R17、R18中任二个可以键合而形成环,此时,形成环的基团满足2n=a+b+c+d+e。另外,R16、R17、R18三个可以键合而形成环,此时,三个中二个基团满足2n=a+b+c+d+e,一个基团满足2n-1=a+b+c+d+e。)上述通式(7)~(9)表示的化学结构中,n优选为0~6的整数,更优选为0~4的整数,特别优选为0~2的整数。应予说明,上述通式(7)~(9)表示的化学结构的R13和R14键合或R16、R17、R18键合而形成环时,n优选为1~8的整数,更优选为1~7的整数,特别优选为1~3的整数。另外,上述通式(7)~(9)表示的化学结构中,a、c、d、e优选为0。金属盐特别优选为(CF3SO2)2NLi(以下有时称为“LiTFSA”)、(FSO2)2NLi(以下有时称为“LiFSA”)、(C2F5SO2)2NLi、FSO2(CF3SO2)NLi、(SO2CF2CF2SO2)NLi、(SO2CF2CF2CF2SO2)NLi、FSO2(CH3SO2)NLi、FSO2(C2F5SO2)NLi、或FSO2(C2H5SO2)NLi。应予说明,这些金属盐为酰亚胺盐。因此,也可以说特别优选使用酰亚胺盐作为金属盐。金属盐采用将以上说明的阳离子和阴离子分别以适当的个数组合而成的金属盐即可。金属盐可以采用上述中的一种,也可以并用多种。另一方面,电解液(1)中的金属盐是在阴离子的化学结构中含有硫元素和氧元素的金属盐,金属盐的阳离子与上述的本发明的电解液相同。电解液(1)中的盐的阴离子的化学结构含有硫元素和氧元素。以下,对该阴离子的化学结构进行具体说明。应予说明,以下仅对本发明的电解液与本发明的电解液(1)不同进行说明。因此,对于没有特别说明的事项,电解液(1)与本发明的电解液相同,盐的阴离子的化学结构优选为上述的通式(1)、通式(2)或通式(3)表示的化学结构,但如下对于X1~X5,与上述的X1~X5相比进一步被限定。电解液(1)中,通式(1)的X1选自SO2、S=O,X2选自SO2、S=O。另外电解液(1)中,通式(2)的X3选自SO2、S=O。另外电解液(1)中,通式(3)的X4选自SO2、S=O,X5选自SO2、S=O,X6选自SO2、S=O。盐的阴离子的化学结构更优选为上述的通式(4)、通式(5)或通式(6)表示的化学结构,但如下对于X7~X12,与上述的X7~X12相比进一步被限定。电解液(1)中,通式(4)的X7选自SO2、S=O,X8选自SO2、S=O。另外,电解液(1)中,通式(5)的X9选自SO2、S=O。另外,电解液(1)中,通式(6)的X10选自SO2、S=O,X11选自SO2、S=O,X12选自SO2、S=O。〔有机溶剂〕作为具有杂元素的有机溶剂,优选杂元素为选自氮、氧、硫、卤素中的至少1个的有机溶剂,更优选杂元素为选自氮或氧中的至少1个的有机溶剂。另外,作为具有杂元素的有机溶剂,优选不具有NH基、NH2基、OH基、SH基等供质子基团的非质子性溶剂。具体例示具有杂元素的有机溶剂(以下,有时简称为“有机溶剂”),可举出乙腈、丙腈、丙烯腈、丙二腈等腈类,1,2-二甲氧基乙烷、1,2-二乙氧基乙烷、四氢呋喃、1,2-二烷、1,3-二烷、1,4-二烷、2,2-二甲基-1,3-二氧戊环、2-甲基四氢吡喃、2-甲基四氢呋喃、冠醚等醚类,碳酸亚乙酯、碳酸亚丙酯、碳酸二甲酯、碳酸二乙酯、碳酸甲乙酯等碳酸酯类,甲酰胺、N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、N-甲基吡咯烷酮等酰胺类,异丙基异氰酸酯、正丙基异氰酸酯、氯甲基异氰酸酯等异氰酸酯类,乙酸甲酯、乙酸乙酯、乙酸丙酯、丙酸甲酯、甲酸甲酯、甲酸乙酯、乙酸乙烯酯、丙烯酸甲酯、甲基丙烯酸甲酯等酯类,缩水甘油基甲醚、环氧丁烷、2-乙基环氧乙烷等环氧类,唑、2-乙基唑、唑啉、2-甲基-2-唑啉等唑类,丙酮、甲乙酮、甲基异丁基酮等酮类,乙酸酐、丙酸酐等酸酐,二甲基砜、环丁砜等砜类,二甲基亚砜等亚砜类,1-硝基丙烷、2-硝基丙烷等硝基类,呋喃、糠醛等呋喃类,γ-丁内酯、γ-戊内酯、δ-戊内酯等环状酯类,噻吩、吡啶等芳香族杂环类,四氢-4-吡喃酮、1-甲基吡咯烷、N-甲基吗啉等杂环类,磷酸三甲酯、磷酸三乙酯等磷酸酯类。此外,作为具有杂元素的有机溶剂,也可举出下述通式(10)表示的链状碳酸酯。R19OCOOR20······通式(10)(R19、R20各自独立地选自为链状烷基的CnHaFbClcBrdIe或化学结构中含有环状烷基的CmHfFgClhBriIj中的任一种。n、a、b、c、d、e、m、f、g、h、i、j各自独立地为0以上的整数,满足2n+1=a+b+c+d+e,2m=f+g+h+i+j。)上述通式(10)表示的链状碳酸酯中,n优选为1~6的整数,更优选为1~4的整数,特别优选为1~2的整数。m优选为3~8的整数,更优选为4~7的整数,特别优选为5~6的整数。另外,上述通式(10)表示的链状碳酸酯中,特别优选碳酸二甲酯(以下有时称为“DMC”)、碳酸二乙酯(以下有时称为“DEC”)、碳酸甲乙酯(以下有时称为“EMC”)。作为具有杂元素的有机溶剂,优选相对介电常数为20以上或具有供给性的醚氧的溶剂,作为这样的有机溶剂,可举出乙腈、丙腈、丙烯腈、丙二腈等腈类,1,2-二甲氧基乙烷、1,2-二乙氧基乙烷、四氢呋喃、1,2-二烷、1,3-二烷、1,4-二烷、2,2-二甲基-1,3-二氧戊环、2-甲基四氢吡喃、2-甲基四氢呋喃、冠醚等醚类,N,N-二甲基甲酰胺、丙酮、二甲基亚砜、环丁砜,特别优选乙腈(以下有时称为“AN”)、1,2-二甲氧基乙烷(以下有时称为“DME”)。这些有机溶剂可以单独用在电解液中,也可以并用多种。本发明的电解液的特征在于,在其振动光谱中,对于来自电解液所含的有机溶剂的峰强度,将有机溶剂本来的峰的强度设为Io、将有机溶剂本来的峰产生位移后的峰(以下有时称为“位移峰”)的强度设为Is时,满足Is>Io。即,在将本发明的电解液供于振动光谱测定而得的振动光谱图表中,上述2个峰强度的关系是Is>Io。这里,“有机溶剂本来的峰”是指在仅对有机溶剂进行振动光谱测定时的峰位置(波数)观察到的峰。有机溶剂本来的峰的强度Io值和位移峰的强度Is值是振动光谱中的各峰距基线的高度或面积。在本发明的电解液的振动光谱中,有机溶剂本来的峰产生位移后的峰存在多个时,基于最容易判断Is和Io的关系的峰判断该关系即可。另外,本发明的电解液使用多种具有杂元素的有机溶剂时,选择最容易判断Is和Io的关系的(Is与Io的差最明显的)有机溶剂,基于其峰强度判断Is和Io的关系即可。另外,峰的位移量小、位移前后的峰重叠而看上去像平缓的山时,可以使用已知的手段进行峰分离,来判定Is和Io的关系。应予说明,在使用了多种具有杂元素的有机溶剂的电解液的振动光谱中,最容易与阳离子配位的有机溶剂(以下有时称为“优先配位溶剂”)的峰优先于其他有机溶剂发生位移。在使用了多种具有杂元素的有机溶剂的电解液中,优先配位溶剂相对于具有杂元素的有机溶剂整体的质量%优选为40%以上,更优选为50%以上,进一步优选为60%以上,特别优选为80%以上。另外,在使用了多种具有杂元素的有机溶剂的电解液中,优先配位溶剂相对于具有杂元素的有机溶剂整体的体积%优选为40%以上,更优选为50%以上,进一步优选为60%以上,特别优选为80%以上。本发明的电解液的振动光谱中的上述2个峰强度的关系优选满足Is>2×Io的条件,更优选满足Is>3×Io的条件,进一步优选满足Is>5×Io的条件,特别优选满足Is>7×Io的条件。最优选的是在本发明的电解液的振动光谱中观察不到有机溶剂本来的峰的强度Io、观察到位移峰的强度Is的电解液。意思是该电解液中电解液所含的有机溶剂的全部分子与金属盐完全溶剂化。最优选的是本发明的电解液为电解液所含的有机溶剂的全部分子与金属盐完全溶剂化的状态(Io=0的状态)。推断本发明的电解液中,金属盐与具有杂元素的有机溶剂(或优先配位溶剂)发生了相互作用。具体而言,推断金属盐与具有杂元素的有机溶剂(或优先配位溶剂)的杂元素形成了配位键,形成了由金属盐和具有杂元素的有机溶剂(或优先配位溶剂)构成的稳定的簇合物(cluster)。从后述的实施例的结果来看,推断该簇合物大致是通过对1分子金属盐配合2分子具有杂元素的有机溶剂(或优先配位溶剂)而形成的。从这点考虑,本发明的电解液中的相对于金属盐1摩尔的具有杂元素的有机溶剂(或优先配位溶剂)的摩尔范围优选为1.4摩尔以上且低于3.5摩尔,更优选为1.5摩尔~3.1摩尔,进一步优选为1.6摩尔~3摩尔。由于推断本发明的电解液中,大致是通过对1分子金属盐配位2分子具有杂元素的有机溶剂(或优先配位溶剂)而形成了簇合物,所以本发明的电解液的浓度(mol/L)依赖于金属盐和有机溶剂各自的分子量和形成溶液时的密度。因此,将本发明的电解液的浓度一概而论是不适当的。表1中分别例示了本发明的电解液的浓度(mol/L)。表1金属盐有机溶剂浓度(mol/L)LiTFSADME2.2~3.4LiTFSAAN3.2~4.9LiFSADME2.6~4.1LiFSAAN3.9~6.0LiFSADMC2.3~4.5LiFSAEMC2.0~3.8LiFSADEC1.8~3.6形成簇合物的有机溶剂和与簇合物形成无关的有机溶剂的各自存在环境不同。因此,振动光谱测定中,来自形成簇合物的有机溶剂的峰被观察到从观察到来自与簇合物形成无关的有机溶剂的峰(有机溶剂本来的峰)的波数向高波数侧或低波数侧位移。即,位移峰相当于形成簇合物的有机溶剂的峰。作为振动光谱,可举出IR光谱或拉曼光谱。作为IR测定的测定方法,可举出石蜡糊法、液膜法等透射测定方法,ATR法等反射测定方法。关于选择IR光谱还是拉曼光谱,只要选择在本发明的电解液的振动光谱中容易判断Is和Io的关系的光谱即可。应予说明,振动光谱测定优选在可以减少或忽略大气中的水分的影响的条件下进行。例如,优选在干燥室、手套箱等低湿度或无湿度条件下进行IR测定或在将电解液放入密闭容器的状态下进行拉曼测定。这里,对含有LiTFSA作为金属盐且含有乙腈作为有机溶剂的本发明的电解液的峰进行具体说明。仅对乙腈进行IR测定时,来自C和N间的三键的伸缩振动的峰通常在2100~2400cm-1附近被观察到。这里,基于现有的技术常识,假定将LiTFSA以1mol/L的浓度溶解于乙腈溶剂而制成电解液的情况。由于乙腈1L相当于约19mol,所以现有的电解液1L中,存在1mol的LiTFSA和19mol的乙腈。这样,现有的电解液中,存在与LiTFSA溶剂化的(与Li配位)乙腈,同时存在大量不与LiTFSA溶剂化的(不与Li配位)乙腈。然而,对于与LiTFSA溶剂化的乙腈分子和不与LiTFSA溶剂化的乙腈分子而言,由于乙腈分子所处的环境不同,所以在IR光谱中,观察到两者的乙腈峰有区别。更具体而言,不与LiTFSA溶剂化的乙腈的峰在与仅对乙腈进行IR测定的情况同样的位置(波数)被观察到,而另一方面,观察到与LiTFSA溶剂化的乙腈的峰的峰位置(波数)向高波数侧位移。而且,现有的电解液的浓度中,由于存在大量不与LiTFSA溶剂化的乙腈,所以在现有的电解液的振动光谱中,乙腈本来的峰的强度Io与乙腈本来的峰产生位移后的峰的强度Is的关系是Is<Io。另一方面,本发明的电解液与现有的电解液相比较,LiTFSA的浓度高,且电解液中与LiTFSA溶剂化的(形成簇合物)乙腈分子数比不与LiTFSA溶剂化的乙腈分子数多。于是,本发明的电解液的振动光谱中的、乙腈本来的峰的强度Io与乙腈本来的峰产生位移后的峰的强度Is的关系是Is>Io。表2中例示了认为对计算本发明的电解液的振动光谱中Io和Is有用的有机溶剂的波数及其归属。应予说明,还包括因振动光谱的测定装置、测定环境、测定条件不同,观察到的峰的波数有时与以下的波数不同的情况。表2有机溶剂波数(cm-1)归属碳酸亚乙酯1769C和O间的双键碳酸亚丙酯1829C和O间的双键乙酸酐1785、1826C和O间的双键丙酮1727C和O间的双键乙腈2285C和N间的三键乙腈899C-C单键DME1099C-O单键DME1124C-O单键N,N-二甲基甲酰胺1708C和O间的双键γ-丁内酯1800C和O间的双键硝基丙烷1563N和O间的双键吡啶977不明二甲基亚砜1017S-O键对于有机溶剂的波数及其归属,可以将公知的数据作为参考。作为参考文献,可举出日本分光学会测定法系列17拉曼分光法,滨口宏夫,平川晓子,学会出版中心,231~249页。另外,通过使用计算机的计算也能够预测认为对Io和Is的计算有用的有机溶剂的波数和有机溶剂与金属盐配位时的波数位移。例如,可以使用Gaussian09(注册商标,Gaussian公司),将密度泛函设为B3LYP,将基底函数设为6-311G++(d,p)来计算。本领域技术人员可以参考表2的记载、公知的数据、计算机的计算结果,选定有机溶剂的峰,计算Io和Is。本发明的电解液与现有的电解液相比较,金属盐和有机溶剂的存在环境不同,且金属盐浓度高,因此可以期待提高电解液中的金属离子输送速度(特别是金属为锂时,提高锂输送速率)、提高电极与电解液界面的反应速度、缓和电池的高倍率充放电时引起的电解液的盐浓度的不均、增大双电层容量等。如下所述,认为这些优异的效果的至少一部分是由来自本发明的电解液形成在负极和/或正极表面的特殊结构的SEI被膜带来的。而且,认为通过该特殊结构的SEI被膜与本发明的电解液的配合,能发挥上述各种优异的效果,例如,提高电极与电解液界面之间的反应速度。此外,本发明的电解液中,由于具有杂元素的有机溶剂的大部分与金属盐形成了簇合物,所以电解液所含的有机溶剂的蒸气压变低。作为其结果,能够减少有机溶剂从本发明的电解液中的挥发。对本发明的电解液的制造方法进行说明。本发明的电解液与现有的电解液相比较,金属盐的含量多,因此在向固体(粉体)的金属盐中添加有机溶剂的制造方法中得到凝聚物,难以制造溶液状态的电解液。因此,本发明的电解液的制造方法中,优选向有机溶剂中缓慢添加金属盐,且边维持电解液的溶液状态边制造。根据金属盐和有机溶剂的种类,本发明的电解液包含金属盐以超过一直以来知道的饱和溶解度的方式溶解于有机溶剂的液体。这样的本发明的电解液的制造方法包括将具有杂元素的有机溶剂和金属盐混合,使金属盐溶解,制备第1电解液的第1溶解工序;在搅拌和/或加温条件下,向上述第1电解液加入上述金属盐,使上述金属盐溶解,制备过饱和状态的第2电解液的第2溶解工序;在搅拌和/或加温条件下,向上述第2电解液加入上述金属盐,使上述金属盐溶解,制备第3电解液的第3溶解工序。这里,上述“过饱和状态”是指在解除了搅拌和/或加温条件的情况或赋予了振动等晶核生成能量的情况下,金属盐晶体从电解液析出的状态。第2电解液是“过饱和状态”,第1电解液和第3电解液不是“过饱和状态”。换言之,本发明的电解液的上述制造方法经过处于热力学稳定的液体状态的包含现有的金属盐浓度的第1电解液,经由热力学不稳定的液体状态的第2电解液,然后成为热力学稳定的新的液体状态的第3电解液,即本发明的电解液。稳定的液体状态的第3电解液在通常的条件下保持液体状态,所以推断在第3电解液中,例如,由相对于1分子锂盐为2分子有机溶剂构成的、靠这些分子间的强的配位键而稳定化的簇合物阻碍锂盐的结晶化。第1溶解工序是将具有杂原子的有机溶剂和金属盐混合,使金属盐溶解,制备第1电解液的工序。为了将具有杂原子的有机溶剂和金属盐混合,可以向具有杂原子的有机溶剂中加入金属盐,也可以向金属盐中加入具有杂原子的有机溶剂。第1溶解工序优选在搅拌和/或加温条件下进行。搅拌速度只要适当地设定即可。加温条件优选用水浴或油浴等恒温槽适当地控制。由于金属盐的溶解时产生溶解热,所以使用受热不稳定的金属盐时,优选严格地控制温度条件。另外,可以预先冷却有机溶剂,也可以在冷却条件下进行第1溶解工序。第1溶解工序和第2溶解工序可以连续实施,也可以暂时保管(静置)第1溶解工序中得到的第1电解液,经过一定时间后,实施第2溶解工序。第2溶解工序是在搅拌和/或加温条件下向第1电解液中加入金属盐,使金属盐溶解,制备过饱和状态的第2电解液的工序。由于第2溶解工序是制备热力学不稳定的过饱和状态的第2电解液,所以必须在搅拌和/或加温条件下进行。可以通过用混合器等带有搅拌器的搅拌装置进行第2溶解工序而处于搅拌条件下,或者可以通过使用搅拌子和使搅拌子工作的装置(搅拌机)进行第2溶解工序而处于搅拌条件下。加温条件优选用水浴或油浴等恒温槽适当地控制。当然,特别优选使用兼具搅拌功能和加温功能的装置或系统进行第2溶解工序。应予说明,这里提及的加温是指将对象物加温至常温(25℃)以上的温度。加温温度更优选为30℃以上,进一步优选为35℃以上。另外,优选加温温度为比有机溶剂的沸点低的温度。第2溶解工序中,加入的金属盐没有充分溶解的情况下,实施搅拌速度的增加和/或进一步的加温。此时,可以向第2溶解工序的电解液加入少量具有杂原子的有机溶剂。如果将第2溶解工序中得到的第2电解液暂时静置则金属盐的晶体析出,因此优选连续实施第2溶解工序和第3溶解工序。第3溶解工序是在搅拌和/或加温条件下向第2电解液中加入金属盐,使金属盐溶解,制备第3电解液的工序。第3溶解工序中,由于需要向过饱和状态的第2电解液中加入金属盐并溶解,所以必须与第2溶解工序同样地在搅拌和/或加温条件下进行。具体的搅拌和/或加温条件与第2溶解工序的条件同样。如果通过第1溶解工序、第2溶解工序和第3溶解工序所添加的有机溶剂与金属盐的摩尔比大体为2:1左右,则第3电解液(本发明的电解液)的制造结束。即便解除搅拌和/或加温条件,金属盐晶体也不会从本发明的电解液析出。从这些情况来看,推断本发明的电解液形成了例如由相对于锂盐1分子为有机溶剂2分子构成的、靠这些分子间的强的配位键而稳定化的簇合物。应予说明,在制造本发明的电解液时,根据金属盐和有机溶剂的种类,在各溶解工序中的处理温度下,即便不经由上述过饱和状态的情况下,使用上述第1~3溶解工序中所述的具体的溶解手段也能够适当地制造本发明的电解液。另外,在制造本发明的电解液的方法中,优选具有对制造中途的电解液进行振动光谱测定的振动光谱测定工序。作为具体的振动光谱测定工序,例如,可以是取样一部分制造中途的各电解液而供于振动光谱测定的方法,也可以是在原位(在场)对各电解液进行振动光谱测定的方法。作为在原位对电解液进行振动光谱测定的方法,可举出向透明的流动池导入制造中途的电解液来测定振动光谱的方法,或者使用透明的制造容器从该容器外进行拉曼测定的方法。通过使本发明的电解液的制造方法包括振动光谱测定工序,能够在制造中途确认电解液中的Is与Io的关系,因此能够判断制造中途的电解液是否变成本发明的电解液,另外,在制造中途的电解液没有变成本发明的电解液时,能够把握追加多少量的金属盐才能使电解液变成本发明的电解液。本发明的电解液中,除上述具有杂元素的有机溶剂以外,还可以加入低极性(低介电常数)或低供给数的、不与金属盐显示特别的相互作用的溶剂,即,对本发明电解液中的上述簇合物的形成和维持没有影响的溶剂。通过将这样的溶剂加入到本发明的电解液中,可以期待在保持本发明的电解液的上述簇合物的形成的状态下降低电解液的粘度的效果。作为不与金属盐显示特别的相互作用的溶剂,具体而言,可例示苯、甲苯、乙苯、邻二甲苯、间二甲苯、对二甲苯、1-甲基萘、己烷、庚烷、环己烷。另外,本发明的电解液中,除上述具有杂元素的有机溶剂以外,还可以加入阻燃性的溶剂。通过将阻燃性的溶剂加入到本发明的电解液中,能够进一步提高本发明的电解液的安全度。作为阻燃性的溶剂,可例示四氯化碳、四氯乙烷、氢氟醚等卤素系溶剂,磷酸三甲酯、磷酸三乙酯等磷酸衍生物。此外,如果将本发明的电解液与聚合物、无机填料混合而形成混合物,则该混合物封入电解液,成为准固体电解质。通过使用准固体电解质作为电池的电解液,能够抑制电池中的电解液的漏液。作为上述聚合物,可以采用在锂离子二次电池等非水电解质二次电池中使用的聚合物、通常的化学交联的聚合物。特别优选聚偏氟乙烯、聚六氟丙烯等可吸收电解液变成凝胶化的聚合物,聚环氧乙烷等向聚合物导入了离子导电性基团的聚合物。作为具体的聚合物,可例示聚丙烯酸甲酯、聚甲基丙烯酸甲酯、聚环氧乙烷、聚环氧丙烷、聚丙烯腈、聚偏氟乙烯、聚乙二醇二甲基丙烯酸酯、聚乙二醇丙烯酸酯、聚缩水甘油(Polyglycidol)、聚四氟乙烯、聚六氟丙烯、聚硅氧烷、聚乙酸乙烯酯、聚乙烯醇、聚丙烯酸、聚甲基丙烯酸、聚衣康酸、聚富马酸、聚巴豆酸、聚当归酸、羧甲基纤维素等聚羧酸,苯乙烯-丁二烯橡胶、腈-丁二烯橡胶、聚苯乙烯、聚碳酸酯、马来酸酐与二元醇类共聚成的不饱和聚酯、具有取代基的聚环氧乙烷衍生物、偏氟乙烯与六氟丙烯的共聚物。另外,作为上述聚合物,可以选择使构成上述具体的聚合物的二种以上的单体共聚而成的共聚物。作为上述聚合物,还优选多糖类。作为具体的多糖类,可例示糖原、纤维素、甲壳质、琼脂糖、卡拉胶、肝素、透明质酸、果胶、支链淀粉、木葡聚糖、直链淀粉。另外,可以采用含有这些多糖类的材料作为上述聚合物,作为该材料,可例示含有琼脂糖等多糖类的琼脂。作为上述无机填料,优选氧化物、氮化物等无机陶瓷。无机陶瓷在其表面具有亲水性和疏水性的官能团。因此,该官能团通过吸引电解液能在无机陶瓷内形成导电性通路。并且,电解液中分散的无机陶瓷利用上述官能团形成无机陶瓷彼此连成的网络,能发挥封入电解液的作用。利用无机陶瓷的这样的功能,能够更恰当地抑制电池中的电解液的漏液。为了恰当地发挥无机陶瓷的上述功能,无机陶瓷优选为粒子形状,特别优选其粒径为纳米水准。作为无机陶瓷的种类,可举出一般的氧化铝、二氧化硅、氧化钛、氧化锆、磷酸锂盐等。另外,无机陶瓷本身可以具有锂传导性,具体而言,可例示Li3N、LiI、LiI-Li3N-LiOH、LiI-Li2S-P2O5、LiI-Li2S-P2S5、LiI-Li2S-B2S3、Li2O-B2S3、Li2O-V2O3-SiO2、Li2O-B2O3-P2O5、Li2O-B2O3-ZnO、Li2O-Al2O3-TiO2-SiO2-P2O5、LiTi2(PO4)3、Li-βAl2O3、LiTaO3。可以采用玻璃陶瓷作为无机填料。玻璃陶瓷能够封入离子性液体,所以对本发明的电解液也可期待同样的效果。作为玻璃陶瓷,可例示xLi2S-(1-x)P2S5表示的化合物、和将该化合物的S的一部分用其它的元素取代而得的化合物以及将该化合物的P的一部分用锗取代而得的化合物。本发明的电解液的密度d(g/cm3)优选为d≥1.2或d≤2.2,更优选为1.2≤d≤2.2的范围内,更优选为1.24≤d≤2.0的范围内,进一步优选为1.26≤d≤1.8的范围内,特别优选为1.27≤d≤1.6的范围内。应予说明,本发明的电解液的密度d(g/cm3)是指在20℃的密度。以下说明的d/c是用上述d除以盐浓度c(mol/L)而得的值。本发明的电解液的d/c为0.15≤d/c≤0.71,优选为0.15≤d/c≤0.56的范围内,更优选为0.25≤d/c≤0.56的范围内,进一步优选为0.26≤d/c≤0.50的范围内,特别优选为0.27≤d/c≤0.47的范围内。本发明的电解液的d/c在特定金属盐和有机溶剂的情况下也可以规定。例如,选择LiTFSA作为金属盐,选择DME作为有机溶剂时,d/c优选为0.42≤d/c≤0.56的范围内,更优选为0.44≤d/c≤0.52的范围内。选择LiTFSA作为金属盐,选择AN作为有机溶剂时,d/c优选为0.35≤d/c≤0.41的范围内,更优选为0.36≤d/c≤0.39的范围内。选择LiFSA作为金属盐,选择DME作为有机溶剂时,d/c优选为0.32≤d/c≤0.46的范围内,更优选为0.34≤d/c≤0.42的范围内。选择LiFSA作为金属盐,选择AN作为有机溶剂时,d/c优选为0.25≤d/c≤0.48的范围内,更优选为0.25≤d/c≤0.38的范围,进一步优选为0.25≤d/c≤0.31的范围内,进一步优选为0.26≤d/c≤0.29的范围内。选择LiFSA作为金属盐,选择DMC作为有机溶剂时,d/c优选为0.32≤d/c≤0.46的范围内,更优选为0.34≤d/c≤0.42的范围内。选择LiFSA作为金属盐,选择EMC作为有机溶剂时,d/c优选为0.34≤d/c≤0.50的范围内,更优选为0.37≤d/c≤0.45的范围内。选择LiFSA作为金属盐,选择DEC作为有机溶剂时,d/c优选为0.36≤d/c≤0.54的范围内,更优选为0.39≤d/c≤0.48的范围内。本发明的电解液与现有的电解液相比较,金属盐和有机溶剂的存在环境不同,密度高,因此可以期待提高电解液中的金属离子输送速度(特别是金属为锂时,提高锂输送速率),提高电极与电解液界面的反应速度,缓和电池的高倍率充放电时引起的电解液的盐浓度的不均,增大双电层容量等。此外,本发明的电解液中,由于密度高,所以电解液所含的有机溶剂的蒸气压变低。作为其结果,能够减少有机溶剂从本发明的电解液的挥发。另外,这样的本发明的电解液的粘度高于现有的电解液的粘度。因此,如果为使用了本发明的电解液的本发明的非水电解质二次电池,即便电池破损,也能抑制电解液泄漏。另外,使用了现有的电解液的非水电解质二次电池在高速充放电循环时容量减少明显。作为其理由之一,认为是由于快速反复充放电时的电解液中产生的Li浓度不均,导致在与电极的反应界面电解液无法供给足够量的Li,也就是说,电解液的Li浓度不均。然而,本发明的电解液的金属浓度相对于现有的电解液而言较高。例如本发明的电解液的优选的Li浓度是一般的电解液的Li浓度的2~5倍左右。认为这样以高浓度含有Li的本发明的电解液中,Li的不均减少,其结果,认为抑制了高速充放电循环时的容量降低。另外,认为由于本发明的电解液为高粘度,所以电极界面的电解液的保液性提高,抑制在电极界面电解液不足的状态(所谓的液枯状态)也是抑制高速充放电循环时的容量降低的一个原因。若对本发明的电解液的粘度η(mPa·s)进行说明,则优选为10<η<500的范围,更优选为12<η<400的范围,进一步优选为15<η<300的范围,特别优选为18<η<150的范围,最优选为20<η<140的范围。另外,电解液的离子传导率σ(mS/cm)越高,离子越容易在电解液中移动。因此,这样的电解液能成为优异的电池的电解液。若对本发明的电解液的离子传导率σ(mS/cm)进行说明,则优选为1≤σ。对于本发明的非水电解质二次电池中的电解液的离子传导率σ(mS/cm),若非要例示包含上限的优选的范围,则优选为2<σ<200的范围,更优选为3<σ<100的范围,进一步优选为4<σ<50的范围,特别优选为5<σ<35的范围。然而,在本发明的非水电解质二次电池(1)的负极和/或正极的表面形成了含S、O被膜。大多情况下,在非水电解质二次电池(2)的负极和/或正极的表面也形成了含S、O被膜。如下所述,该被膜含有S和O,至少具有S=O结构。而且,由于该含S、O被膜具有S=O结构,所以认为是来自电解液的。认为在本发明的电解液中,与通常的电解液相比,Li阳离子和阴离子离得较近。因此阴离子受来自Li阳离子的静电影响强而优先被还原分解。在使用了一般的电解液的一般的非水电解质二次电池中,电解液所含的有机溶剂(例如EC:碳酸亚乙酯等)被还原分解,由该有机溶剂的分解产物构成SEI被膜。但是,如上所述本发明的非水电解质二次电池所含的本发明的电解液中阴离子优先被还原分解。因此,认为本发明的非水电解质二次电池中的SEI被膜即含S、O被膜含有大量来自阴离子的S=O结构。也就是说,在使用了通常的电解液的通常的非水电解质二次电池中,来自EC等有机溶剂的分解物的SEI被膜固定在电极表面。另一方面,在使用了本发明的电解液的本发明的非水电解质二次电池中,主要是来自金属盐的阴离子的SEI被膜固定在电极表面。另外,虽理由尚不确定,但本发明的非水电解质二次电池中的含S、O被膜伴随充放电发生状态变化。例如,如下所述,根据充放电的状态含S、O被膜的厚度、S、O等元素的比例有时变化。因此,认为本发明的非水电解质二次电池的含S、O被膜中存在来自上述阴离子的分解物、固定在被膜中的部分(以下,根据需要称为固定部)和伴随充放电可逆地增减的部分(以下,根据需要称为吸附部)。而且推测吸附部与固定部同样地具有来自金属盐的阴离子的S=O等结构。应予说明,认为含S、O被膜由电解液的分解物构成,另外含有吸附物,因此认为含S、O被膜的大部分(或全部)是在非水电解质二次电池的初次充放电以后生成的。也就是说,本发明的非水电解质二次电池在使用时在负极的表面和/或正极的表面具有含S、O被膜。含S、O被膜的其它的构成成分根据电解液所含的硫和氧以外的成分、负极的组成等各有不同。另外,该含S、O被膜只要含有S=O结构即可,其含有比例没有特别限定。此外,含S、O被膜所含的S=O结构以外的成分和量没有特别限定。而且,含S、O被膜可以仅在负极表面形成,也可以仅在正极表面形成。然而,如上所述认为含S、O被膜是来自本发明的电解液所含的金属盐的阴离子,因此优选来自该金属盐的阴离子的成分比其它的成分含有的多。另外,优选含S、O被膜在负极表面和正极表面形成。以下,根据需要,将在负极的表面形成的含S、O被膜称为负极含S、O被膜,将在正极的表面形成的含S、O被膜称为正极含S、O被膜。如上所述,作为本发明的电解液中的金属盐,可优选使用酰亚胺盐。一直以来,已知有向电解液中添加酰亚胺盐的技术,知道在使用了这种电解液的非水电解质二次电池中,正极和/或负极上的被膜除含有来自电解液的有机溶剂分解物的化合物以外,还含有来自酰亚胺盐的化合物即含S的化合物。例如日本特开2013-145732中,介绍了利用该被膜含有的一部分来自酰亚胺盐的成分,能抑制非水电解质二次电池的内部电阻的增大,并且能提高非水电解质二次电池的耐久性。然而,上述的现有技术中,由于以下的理由被膜中的来自酰亚胺盐的成分无法浓化。首先,使用石墨作为负极活性物质时,为了使石墨与电荷载体发生可逆性反应,使非水电解质二次电池可逆地进行充放电,认为需要在负极的表面形成SEI被膜。以往,为了形成该SEI被膜,使用以EC为代表的环状碳酸酯化合物作为电解液用的有机溶剂。而且,利用该环状碳酸酯化合物的分解物形成了SEI被膜。也就是说,含有酰亚胺盐的现有电解液含有大量作为有机溶剂的EC等环状碳酸酯且含有作为添加剂的酰亚胺盐。但是,此时,SEI被膜的主成分是来自有机溶剂的成分,难以使SEI被膜的酰亚胺盐的含量增多。另外,想要将酰亚胺盐不作为添加剂而作为金属盐(也就是说电解质盐、支持盐)使用时,必须考虑与正极用的集电体的组合。也就是说,已知酰亚胺盐腐蚀通常作为正极用集电体使用的铝集电体。因此,特别是使用以4V左右的电位工作的正极时,需要使以跟铝形成钝化物的LiPF6等为电解质盐的电解液与铝集电体共存。另外,在现有的电解液中,从离子传导率、粘度的观点考虑,由LiPF6、酰亚胺盐等构成的电解质盐的合计浓度最佳为1mol/L~2mol/L左右(日本特开2013-145732)。因此如果添加足够量的LiPF6,则酰亚胺盐的添加量必然减少,所以存在难以大量使用酰亚胺盐作为电解液用的金属盐的问题。以下,根据需要,有时将酰亚胺盐简称为金属盐。与此相对,本发明的电解液以高浓度含有金属盐。而且,如下所述,认为本发明的电解液中金属盐以与以往完全不同的状态存在。因此,本发明的电解液与现有的电解液不同,不易出现因金属盐为高浓度而产生的问题。例如,利用本发明的电解液,能够抑制由电解液的粘度上升引起的非水电解质二次电池的输入输出性能降低,还能够抑制铝集电体的腐蚀。另外,电解液以高浓度含有的金属盐在负极上优先被还原分解。其结果,即便不使用作为有机溶剂的EC等环状碳酸酯化合物,也能在负极上形成来自金属盐的特殊结构的SEI被膜,即含S、O被膜。因此本发明的非水电解质二次电池即便使用石墨作为负极活性物质时,也能够在有机溶剂不使用环状碳酸酯化合物的情况下可逆地进行充放电。因此本发明的非水电解质二次电池即便使用石墨作为负极活性物质且使用铝集电体作为正极用集电体时,也能够在不使用作为有机溶剂的环状碳酸酯化合物或者作为金属盐的LiPF6的情况下可逆地进行充放电。并且能够使负极和/或正极表面的SEI被膜的大部分由来自阴离子的成分构成。如下所述,利用含有来自阴离子的成分的含S、O被膜能提高非水电解质二次电池的电池特性。应予说明,使用了含有EC溶剂的一般电解液的非水电解质二次电池中负极的被膜含有大量来自EC溶剂的碳聚合而成的聚合物结构。与此相对,本发明的非水电解质二次电池中的负极含S、O被膜几乎(或完全)不含这样的碳聚合而成的聚合物结构,而含有大量来自金属盐的阴离子的结构。正极被膜也是同样的。然而,本发明的电解液以高浓度含有金属盐的阳离子。因此,本发明的电解液中,相邻的阳离子间的距离极近。而且,在非水电解质二次电池的充放电时,锂离子等阳离子在正极与负极之间移动时,与移动目标的电极最近的阳离子首先被供给给该电极。然后,在被供给的该阳离子存在过的场所,与该阳离子相邻的其它的阳离子移动。也就是说,预料本发明的电解液中,产生相邻的阳离子朝向作为供给对象的电极依次一个一个地改变位置这样的如多米诺骨牌似的现象。因此,认为充放电时的阳离子的移动距离短,仅仅这样阳离子的移动速度就高。而且,由此,认为具有本发明的电解液的本发明的非水电解质二次电池的反应速度高。另外,认为本发明的非水电解质二次电池的电极(也就是说负极和/或正极)具有含S、O被膜,该含S、O被膜具有S=O结构且含有大量的阳离子。认为该含S、O被膜所含的阳离子优先被供给到电极。因此,认为本发明的非水电解质二次电池中,由于电极附近具有丰富的阳离子源(也就是说含S、O被膜)而进一步提高阳离子的输送速度。因此,认为本发明的非水电解质二次电池中,通过本发明的电解液与含S、O被膜的配合,发挥优异的电池特性。仅限于参考,认为负极的SEI被膜是电解液在规定以下的电压下还原分解,由此时生成的电解液的堆积物构成的。也就是说,为了在负极的表面高效生成上述的含S、O被膜,优选使本发明的非水电解质二次电池的负极的电位的最小值达到规定以下。具体而言,本发明的非水电解质二次电池在对电极为锂时优选为在负极的电位的最小值为1.3V以下的条件下使用的电池。本发明的非水电解质二次电池中的负极没有特别限定。作为负极活性物质,可使用能吸留和放出电荷载体的一般的负极活性物质。例如,非水电解质二次电池为锂离子二次电池时,作为负极活性物质,只要选择能吸留和放出锂离子的材料即可。更详细而言,可以为能与Li等电荷载体发生合金化的元素(单质)、含有该元素的合金或含有该元素的化合物。具体而言,作为负极活性物质,可以以单质的形式分别采用Li、碳、硅、锗、锡等第14族元素,铝、铟等第13族元素,锌、镉等第12族元素,锑、铋等第15族元素,镁、钙等碱土金属,银、金等第11族元素。如果负极活性物质采用硅等,则由于1个硅原子与多个锂反应,所以是高容量的活性物质,但由于可能出现伴随锂的吸留和放出产生的体积的膨胀和收缩明显的问题,所以为了减少这种可能性,还优选采用硅等单质与过渡金属等其它的元素组合成的合金或化合物作为负极活性物质。作为合金或化合物的具体例,可举出Ag-Sn合金、Cu-Sn合金、Co-Sn合金等锡系材料,各种石墨等碳系材料,歧化成硅单质与二氧化硅的SiOx(0.3≤x≤1.6)等硅系材料,硅单质或硅系材料与碳系材料组合成的复合体。另外,作为负极活性物质,也可以采用Nb2O5、TiO2、Li4Ti5O12、WO2、MoO2、Fe2O3等氧化物,或Li3-xMxN(M=Co、Ni、Cu)表示的氮化物。作为负极活性物质,可以使用这些物质的一种以上。如上所述,本发明的非水电解质二次电池(1)在负极表面形成了含S、O被膜。因此,能够应对低电位负极。具体而言,非水电解质二次电池(1)可选择石墨等含有碳元素的物质、Si系的负极活性物质作为负极活性物质。石墨可以为天然石墨、人造石墨,其粒径也没有特别限定。本发明的非水电解质二次电池具备:具有能吸留和放出锂离子等电荷载体的负极活性物质的负极;具有能吸留和放出该电荷载体的正极活性物质的正极;和上述的本发明的电解液。例如,本发明的非水电解质二次电池为锂离子二次电池时,负极活性物质能吸留和放出锂离子,正极活性物质能吸留和放出锂离子,电解液采用锂盐作为金属盐。负极具有集电体和粘结在集电体的表面的负极活性物质层。关于负极活性物质,已经叙述了。集电体是指用于在非水电解质二次电池的放电或充电时对电极持续流通电流的化学性稳定的电子高传导体。作为负极用的集电体,可例示选自银、铜、金、铝、钨、钴、锌、镍、铁、铂、锡、铟、钛、钌、钽、铬、钼中的至少一种以及不锈钢等金属材料。集电体可以用公知的保护层被覆。可以使用将集电体的表面用公知方法处理了的集电体作为集电体。集电体可以采用箔片、薄片、膜、线状、棒状、网等形态。因此,作为集电体,例如,可优选使用铜箔、镍箔、铝箔、不锈钢箔等金属箔。集电体为箔片、薄片、膜的形态时,其厚度优选为1μm~100μm的范围内。负极活性物质层包括负极活性物质以及根据需要的粘结剂和/或导电助剂。非水电解质二次电池(2)使用特定的粘结剂。粘结剂起到使负极活性物质粒子之间或者负极活性物质和导电助剂固定在集电体的表面的作用。非水电解质二次电池(2)的粘结剂含有具有亲水基团的聚合物。作为具有亲水基团的聚合物的亲水基团,可例示羧基、磺基、硅烷醇基、氨基、羟基、氨基、磷酸基等磷酸系的基团等。其中,优选聚丙烯酸(PAA)、羧甲基纤维素(CMC)、聚甲基丙烯酸等分子中含有羧基的聚合物,或者聚(对苯乙烯磺酸)等含有磺基的聚合物。聚丙烯酸或者丙烯酸与乙烯基磺酸的共聚物等含有大量羧基和/或磺基的聚合物为水溶性。因此具有亲水基团的聚合物优选为水溶性聚合物,优选为一分子中含有多个羧基和/或磺基的聚合物。分子中含有羧基的聚合物例如可以用将聚丙烯酸等酸单体聚合的方法、或者对羧甲基纤维素(CMC)等聚合物赋予羧基的方法等方法来制造。作为酸单体,可例示丙烯酸、甲基丙烯酸、乙烯基苯甲酸、巴豆酸、戊烯酸、当归酸、顺芷酸等分子中具有一个羧基的酸单体;衣康酸、中康酸、柠康酸、富马酸、马来酸、2-戊烯二酸、亚甲基琥珀酸、烯丙基丙二酸、亚异丙基琥珀酸、2,4-己二烯二酸、乙炔二羧酸等分子内具有二个以上的羧基的酸单体等。可以使用将从这些中选择的二种以上的单体进行聚合而成的共聚聚合物。还优选使用例如如日本特开2013-065493号公报所述的由丙烯酸和衣康酸的共聚物构成的、分子中含有羧基彼此缩合而形成的酸酐基的聚合物作为粘结剂。认为通过具有来自一分子中具有二个以上的羧基的酸性度高的单体的结构,在充电时在发生电解液分解反应前容易捕捉锂离子等。此外,由于与聚丙烯酸、聚甲基丙烯酸相比羧基多、酸性度高,并且规定量的羧基变化成酸酐基,所以酸性度也不会过高。因此,具有使用该负极用粘结剂形成的负极的二次电池,其初始效率提高,输入输出特性提高。另外在不损害性能的范围内,可以混合聚偏氟乙烯、聚四氟乙烯、氟橡胶等含氟树脂,聚丙烯、聚乙烯等热塑性树脂,聚酰亚胺、聚酰胺酰亚胺等酰亚胺系树脂,含有烷氧基甲硅烷基的树脂等聚合物。负极活性物质层中的粘结剂的配合比例以质量比计,优选为负极活性物质:粘结剂=1:0.005~1:0.3。这是由于如果粘结剂过少则电极的成型性降低,另外,如果粘结剂过多则电极的能量密度变低。非水电解质二次电池(1)的粘结剂可以为上述的粘结剂,也可以为其它的粘结剂。例如,作为粘结剂,可例示聚偏氟乙烯、聚四氟乙烯、氟橡胶等含氟树脂,聚丙烯、聚乙烯等热塑性树脂,聚酰亚胺、聚酰胺酰亚胺等酰亚胺系树脂,含有烷氧基甲硅烷基的树脂。任何情况下,负极活性物质层中的粘结剂的配合比例以质量比计,均优选为负极活性物质:粘结剂=1:0.005~1:0.3。这是由于如果粘结剂过少则电极的成型性降低,另外,如果粘结剂过多则电极的能量密度变低。导电助剂是为了提高电极的导电性而添加的。因此,导电助剂在电极的导电性不足的情况下可以任意添加,在电极的导电性足够优异的情况下可以不添加。作为导电助剂,只要为化学性稳定的电子高传导体即可,可例示为碳质微粒的炭黑、石墨、乙炔黑、科琴黑(注册商标)、气相生长碳纤维(VaporGrownCarbonFiber:VGCF)和各种金属粒子等。这些导电助剂可以单独或组合二种以上添加到活性物质层。负极活性物质层中的导电助剂的配合比例以质量比计,优选为负极活性物质:导电助剂=1:0.01~1:0.5。这是由于如果导电助剂过少则无法形成高效率的导电通道,另外,如果导电助剂过多则负极活性物质层的成型性差且电极的能量密度变低。使用上述粘结剂,制作非水电解质二次电池的负极时,可以通过将加入负极活性物质粉末、碳粉末等导电助剂、上述粘结剂和适量的溶剂并混合而制成浆料的物质用辊涂法、浸涂法、刮刀法、喷涂法、帘涂法等方法涂布在集电体上,使上述粘结剂干燥或者固化而制作。作为溶剂,可例示N-甲基-2-吡咯烷酮、甲醇、甲基异丁基酮、水。为了提高电极密度,可以将干燥后的物质进行压缩。〔正极〕非水电解质二次电池中使用的正极具有能吸留和放出锂离子等电荷载体的正极活性物质。正极具有集电体和粘结在集电体的表面的正极活性物质层。正极活性物质层包括正极活性物质以及根据需要的粘结剂和/或导电助剂。正极的集电体只要为可耐受适合所使用的活性物质的电压的金属就没有特别限制,例如,可例示选自银、铜、金、铝、钨、钴、锌、镍、铁、铂、锡、铟、钛、钌、钽、铬、钼中的至少一种以及不锈钢等金属材料。正极的电位以锂基准计为4V以上时,优选采用铝作为集电体。具体而言,作为正极用集电体,优选使用由铝或铝合金构成的集电体。这里铝是指纯铝,将纯度99.0%以上的铝称为纯铝。将向纯铝中添加各种元素而成为合金的物质称为铝合金。作为铝合金,可举出Al-Cu系、Al-Mn系、Al-Fe系、Al-Si系、Al-Mg系、AL-Mg-Si系、Al-Zn-Mg系。另外,作为铝或铝合金,具体而言,例如可举出JISA1085、A1N30等A1000系合金(纯铝系),JISA3003、A3004等A3000系合金(Al-Mn系),JISA8079、A8021等A8000系合金(Al-Fe系)。集电体可以用公知的保护层被覆。可以使用将集电体的表面用公知的方法处理了的集电体作为集电体。集电体可以采用箔片、薄片、膜、线状、棒状、网等形态。因此,作为集电体,例如,可优选使用铜箔、镍箔、铝箔、不锈钢箔等金属箔。集电体为箔片、薄片、膜的形态时,其厚度优选为1μm~100μm的范围内。这与上述的负极用的集电体是同样的。正极的粘结剂和导电助剂与负极中说明的粘结剂和导电助剂同样。作为正极活性物质,可举出层状化合物的LiaNibCocMndDeOf(0.2≤a≤1.2,b+c+d+e=1,0≤e<1,D为选自Li、Fe、Cr、Cu、Zn、Ca、Mg、S、Si、Na、K、Al、Zr、Ti、P、Ga、Ge、V、Mo、Nb、W、La中的至少1种元素,1.7≤f≤2.1)、Li2MnO3。另外,作为正极活性物质,可举出LiMn2O4等尖晶石,以及由尖晶石和层状化合物的混合物构成的固溶体,由LiMPO4、LiMVO4或Li2MSiO4(式中的M选自Co、Ni、Mn、Fe中的至少一种)等表示的聚阴离子系化合物。此外,作为正极活性物质,可举出LiFePO4F等由LiMPO4F(M为过渡金属)表示的羟磷铁锂(Tavorite)系化合物、LiFeBO3等由LiMBO3(M为过渡金属)表示的硼酸盐系化合物。作为正极活性物质使用的任何金属氧化物均可以将上述的组成式作为基本组成,也可以使用将基本组成所含的金属元素用其它的金属元素取代而成的物质。另外,作为正极活性物质,可以使用不含电荷载体(例如有助于充放电的锂离子)的正极活性物质。例如,也可以使用硫单质(S)、硫与碳复合化的化合物、TiS2等金属硫化物、V2O5、MnO2等氧化物、聚苯胺和蒽醌以及化学结构中含有这些芳香族的化合物、共轭二乙酸系有机物等共轭系材料、其它公知的材料。此外,可以采用具有硝基氧(nitroxide)、氮氧自由基(nitronylnitroxide)、加尔万氧自由基(galvinoxyl)、苯氧基等稳定的自由基的化合物作为正极活性物质。使用不含锂等电荷载体的正极活性物质材料时,需要利用公知的方法向正极和/或负极预先添加电荷载体。电荷载体可以以离子的状态添加,可以以金属等非离子的状态添加。例如,电荷载体为锂时,可以将锂箔贴附于正极和/或负极等将其一体化。正极与负极同样地可以含有导电助剂和粘结剂等。导电助剂和粘结剂没有特别限定,与上述的负极同样地,只要是能够用在非水电解质二次电池中的导电助剂和粘结剂即可。在集电体的表面形成活性物质层时,可以使用辊涂法、模涂法、浸涂法、刮刀法、喷涂法、帘涂法等一直以来公知的方法,在集电体的表面涂布活性物质。具体而言,制备含有活性物质以及根据需要的粘结剂和导电助剂的活性物质层形成用组合物(所谓负极合材、正极合材),向该组合物加入适当的溶剂制成糊状后,涂布在集电体的表面后,进行干燥。作为溶剂,可例示N-甲基-2-吡咯烷酮、甲醇、甲基异丁基酮、水。为了提高电极密度,可以将干燥后的物品进行压缩。非水电解质二次电池根据需要使用隔离件。隔离件隔离正极与负极,防止因两极的接触导致的电流的短路,并且使锂离子通过。作为隔离件,可举出使用了聚四氟乙烯、聚丙烯、聚乙烯、聚酰亚胺、聚酰胺、芳族聚酰胺(Aromaticpolyamide)、聚酯、聚丙烯腈等合成树脂、纤维素、直链淀粉等多糖类、丝心蛋白、角蛋白、木质、软木脂等天然高分子、陶瓷等电绝缘性材料中1种或多种的多孔体、无纺布、织物等。另外,隔离件可以为多层结构。由于本发明的电解液的粘度稍高,极性高,所以优选水等极性溶剂容易浸入的膜。具体而言,进一步优选水等极性溶剂浸入存在的空隙的90%以上的膜。在正极与负极之间根据需要夹设隔离件而制成电极体。电极体可以是正极、隔离件和负极重叠成的层叠型或正极、隔离件和负极卷成的卷绕型中的任一类型。在将正极的集电体和负极的集电体到连通外部的正极端子和负极端子之间使用集电用导线等连接后,向电极体加入本发明的电解液可以制成非水电解质二次电池。另外,本发明的非水电解质二次电池只要在适合电极所含的活性物质的种类的电压范围执行充放电即可。本发明的非水电解质二次电池的形状没有特别限定,可以采用圆柱型、方型、纽扣型、层压型等各种形状。本发明的非水电解质二次电池如上所述与电荷载体的种类无关。因此,本发明的非水电解质二次电池例如可以为锂离子二次电池,可以为锂二次电池。或者,可以使用锂以外的电荷载体(例如钠)。本发明的非水电解质二次电池可以搭载于车辆。车辆为其动力源的全部或者一部份使用非水电解质二次电池产生的电能的车辆即可,例如,电动汽车、混合动力汽车等。在车辆搭载非水电解质二次电池时,可以将多个非水电解质二次电池以串联的方式连接而形成电池组。作为搭载非水电解质二次电池的设备,除车辆以外,还可举出个人计算机、便携式通信设备等用电池驱动的各种家电产品、办公设备、产业设备等。此外,本发明的非水电解质二次电池可以用于风力发电、太阳能发电、水力发电及其它电力系统的蓄电装置和电力平滑化装置、船舶等的动力和/或辅助机械类的电力供给源、飞机、航天器等的动力和/或辅助机械类的电力供给源、动力源不使用电的车辆的辅助用电源、移动式的家庭用机器人的电源、系统备用电源、不间断电源装置的电源、电动汽车用充电站等暂时储存充电所需的电力的蓄电装置。以上,说明了本发明的电解液的实施方式,但本发明不限于上述实施方式。在不脱离本发明的主旨的范围内,能以实施了本领域技术人员可进行的变更、改进等的各种方式来实施。以下,示出实施例和比较例,对本发明进行具体说明。应予说明,本发明不限于这些实施例。以下,只要没有特别说明,则“份”表示质量份,“%”表示质量%。〔电解液〕(E1)如下制造本发明的电解液。将作为有机溶剂的1,2-二甲氧基乙烷约5mL放入具备搅拌子和温度计的烧瓶中。在搅拌条件下,以溶液温度保持在40℃以下的方式向上述烧瓶中的1,2-二甲氧基乙烷缓慢地添加作为锂盐的(CF3SO2)2NLi使其溶解。由于在加入约13g的(CF3SO2)2NLi的时刻(CF3SO2)2NLi的溶解暂时停滞,所以将上述烧瓶投入恒温槽,将烧瓶内的溶液温度加温至50℃,使(CF3SO2)2NLi溶解。由于在加入约15g的(CF3SO2)2NLi的时刻(CF3SO2)2NLi的溶解再次停滞,所以用滴管滴加1滴1,2-二甲氧基乙烷,之后(CF3SO2)2NLi溶解。进一步缓慢地添加(CF3SO2)2NLi,加入全部规定的(CF3SO2)2NLi。将得到的电解液移至20mL容量瓶,加入1,2-二甲氧基乙烷直至容积变成20mL。将其作为电解液E1。得到的电解液的容积为20mL,该电解液所含的(CF3SO2)2NLi为18.38g。电解液E1中的(CF3SO2)2NLi的浓度为3.2mol/L。电解液E1中,相对于1分子(CF3SO2)2NLi含有1,2-二甲氧基乙烷1.6分子。应予说明,上述制造是在非活性气体环境下的手套箱内进行的。(E2)使用16.08g的(CF3SO2)2NLi,用与E1同样的方法,制造(CF3SO2)2NLi的浓度为2.8mol/L的电解液E2。电解液E2中,相对于1分子(CF3SO2)2NLi含有1,2-二甲氧基乙烷2.1分子。(E3)将作为有机溶剂的乙腈约5mL放入具备搅拌子的烧瓶中。在搅拌条件下,向上述烧瓶中的乙腈缓慢地添加作为锂盐的(CF3SO2)2NLi,使其溶解。加入总量为19.52g的(CF3SO2)2NLi后,搅拌一晚。将得到的电解液移至20mL容量瓶,加入乙腈直至容积变成20mL。将其作为电解液E3。应予说明,上述制造是在非活性气体环境下的手套箱内进行的。电解液E3中的(CF3SO2)2NLi的浓度为3.4mol/L。电解液E3中,相对于1分子(CF3SO2)2NLi含有乙腈3分子。(E4)使用24.11g的(CF3SO2)2NLi,用与E3同样的方法,制造(CF3SO2)2NLi的浓度为4.2mol/L的电解液E4。电解液E4中,相对于1分子(CF3SO2)2NLi含有乙腈1.9分子。(E5)使用13.47g的(FSO2)2NLi作为锂盐,使用1,2-二甲氧基乙烷作为有机溶剂,除此之外,用与E3同样的方法,制造(FSO2)2NLi的浓度为3.6mol/L的电解液E5。电解液E5中,相对于1分子(FSO2)2NLi含有1,2-二甲氧基乙烷1.9分子。(E6)使用14.97g的(FSO2)2NLi,用与E5同样的方法,制造(FSO2)2NLi的浓度为4.0mol/L的电解液E6。电解液E6中,相对于1分子(FSO2)2NLi含有1,2-二甲氧基乙烷1.5分子。(E7)使用15.72g的(FSO2)2NLi作为锂盐,除此之外,用与E3同样的方法,制造(FSO2)2NLi的浓度为4.2mol/L的电解液E7。电解液E7中,相对于1分子(FSO2)2NLi含有乙腈3分子。(E8)使用16.83g的(FSO2)2NLi,用与E7同样的方法,制造(FSO2)2NLi的浓度为4.5mol/L的电解液E8。电解液E8中,相对于1分子(FSO2)2NLi含有乙腈2.4分子。(E9)使用18.71g的(FSO2)2NLi,用与E7同样的方法,制造(FSO2)2NLi的浓度为5.0mol/L的电解液E9。电解液E9中,相对于1分子(FSO2)2NLi含有乙腈2.1分子。(E10)使用20.21g的(FSO2)2NLi,用与E7同样的方法,制造(FSO2)2NLi的浓度为5.4mol/L的电解液E10。电解液E10中,相对于1分子(FSO2)2NLi含有乙腈2分子。(E11)将作为有机溶剂的碳酸二甲酯约5mL加入具备搅拌子的烧瓶中。在搅拌条件下,向上述烧瓶中的碳酸二甲酯缓慢地添加作为锂盐的(FSO2)2NLi,使其溶解。加入总量为14.64g的(FSO2)2NLi后,搅拌一晚。将得到的电解液移至20mL容量瓶,加入碳酸二甲酯直至容积变成20mL。将其作为电解液E11。应予说明,上述制造是在非活性气体环境下的手套箱内进行的。电解液E11中的(FSO2)2NLi的浓度为3.9mol/L。电解液E11中,相对于1分子(FSO2)2NLi含有碳酸二甲酯2分子。(E12)向电解液E11加入碳酸二甲酯进行稀释,制成(FSO2)2NLi的浓度为3.4mol/L的电解液E12。电解液E12中,相对于1分子(FSO2)2NLi含有碳酸二甲酯2.5分子。(E13)向电解液E11加入碳酸二甲酯进行稀释,制成(FSO2)2NLi的浓度为2.9mol/L的电解液E13。电解液E13中,相对于1分子(FSO2)2NLi含有碳酸二甲酯3分子。(E14)向电解液E11加入碳酸二甲酯进行稀释,制成(FSO2)2NLi的浓度为2.6mol/L的电解液E14。电解液E14中,相对于1分子(FSO2)2NLi含有碳酸二甲酯3.5分子。(E15)向电解液E11加入碳酸二甲酯进行稀释,制成(FSO2)2NLi的浓度为2.0mol/L的电解液E15。电解液E15中,相对于1分子(FSO2)2NLi含有碳酸二甲酯5分子。(E16)将作为有机溶剂的碳酸甲乙酯约5mL加入具备搅拌子的烧瓶中。在搅拌条件下,向上述烧瓶中的碳酸甲乙酯缓慢地添加作为锂盐的(FSO2)2NLi,使其溶解。加入总量为12.81g的(FSO2)2NLi后,搅拌一晚。将得到的电解液移至20mL容量瓶,加入碳酸甲乙酯直至容积变成20mL。将其作为电解液E16。应予说明,上述制造是在非活性气体环境下的手套箱内进行的。电解液E16中的(FSO2)2NLi的浓度为3.4mol/L。电解液E16中,相对于1分子(FSO2)2NLi含有碳酸甲乙酯2分子。(E17)向电解液E16加入碳酸甲乙酯进行稀释,制成(FSO2)2NLi的浓度为2.9mol/L的电解液E17。电解液E17中,相对于1分子(FSO2)2NLi含有碳酸甲乙酯2.5分子。(E18)向电解液E16加入碳酸甲乙酯进行稀释,制成(FSO2)2NLi的浓度为2.2mol/L的电解液E18。电解液E18中,相对于1分子(FSO2)2NLi含有碳酸甲乙酯3.5分子。(E19)将作为有机溶剂的碳酸二乙酯约5mL加入具备搅拌子的烧瓶中。在搅拌条件下,向上述烧瓶中的碳酸二乙酯缓慢地添加作为锂盐的(FSO2)2NLi,使其溶解。加入总量为11.37g的(FSO2)2NLi后,搅拌一晚。将得到的电解液移至20mL容量瓶,加入碳酸二乙酯直至容积变成20mL。将其作为电解液E19。应予说明,上述制造是在非活性气体环境下的手套箱内进行的。电解液E19中的(FSO2)2NLi的浓度为3.0mol/L。电解液E19中,相对于1分子(FSO2)2NLi含有碳酸二乙酯2分子。(E20)向电解液E19加入碳酸二乙酯进行稀释,制成(FSO2)2NLi的浓度为2.6mol/L的电解液E20。电解液E20中,相对于1分子(FSO2)2NLi含有碳酸二乙酯2.5分子。(E21)向电解液E19加入碳酸二乙酯进行稀释,制成(FSO2)2NLi的浓度为2.0mol/L的电解液E21。电解液E21中,相对于1分子(FSO2)2NLi含有碳酸二乙酯3.5分子。(C1)使用5.74g的(CF3SO2)2NLi,使用1,2-二甲氧基乙烷作为有机溶剂,除此之外,用与E3同样的方法,制造(CF3SO2)2NLi的浓度为1.0mol/L的电解液C1。电解液C1中,相对于1分子(CF3SO2)2NLi含有1,2-二甲氧基乙烷8.3分子。(C2)使用5.74g的(CF3SO2)2NLi,用与E3同样的方法,制造(CF3SO2)2NLi的浓度为1.0mol/L的电解液C2。电解液C2中,相对于1分子(CF3SO2)2NLi含有乙腈16分子。(C3)使用3.74g的(FSO2)2NLi,用与E5同样的方法,制造(FSO2)2NLi的浓度为1.0mol/L的电解液C3。电解液C3中,相对于1分子(FSO2)2NLi含有1,2-二甲氧基乙烷8.8分子。(C4)使用3.74g的(FSO2)2NLi,用与E7同样的方法,制造(FSO2)2NLi的浓度为1.0mol/L的电解液C4。电解液C4中,相对于1分子(FSO2)2NLi含有乙腈17分子。(C5)使用碳酸亚乙酯和碳酸二乙酯的混合溶剂(体积比3:7,以下有时称为“EC/DEC”)作为有机溶剂,使用3.04g的LiPF6作为锂盐,除此之外,用与E3同样的方法,制造LiPF6的浓度为1.0mol/L的电解液C5。(C6)向电解液E11加入碳酸二甲酯进行稀释,制成(FSO2)2NLi的浓度为1.1mol/L的电解液C6。电解液C6中,相对于1分子(FSO2)2NLi含有碳酸二甲酯10分子。(C7)向电解液E16加入碳酸甲乙酯进行稀释,制成(FSO2)2NLi的浓度为1.1mol/L的电解液C7。电解液C7中,相对于1分子(FSO2)2NLi含有碳酸甲乙酯8分子。(C8)向电解液E19加入碳酸二乙酯进行稀释,制成(FSO2)2NLi的浓度为1.1mol/L的电解液C8。电解液C8中,相对于1分子(FSO2)2NLi含有碳酸二乙酯7分子。表3中示出电解液的一览表。表3LiTFSA:(CF3SO2)2NLi、LiFSA:(FSO2)2NLi、AN:乙腈、DME:1,2-二甲氧基乙烷、EC/DTC:碳酸亚乙酯和碳酸二乙酯的混合溶剂(体积比3∶7)(评价例1:IR测定)对电解液E3、E4、E7、E8、E10、C2、C4以及乙腈、(CF3SO2)2NLi、(FSO2)2NLi,按以下的条件进行IR测定。将2100~2400cm-1的范围的IR光谱分别示于图1~图10。图的横轴为波数(cm-1),纵轴为吸光度(反射吸光度)。进一步对电解液E11~E21、电解液C6~C8以及碳酸二甲酯、碳酸甲乙酯、碳酸二乙酯,按以下的条件进行IR测定。将1900~1600cm-1的范围的IR光谱分别示于图11~图27。另外,针对(FSO2)2NLi,将1900~1600cm-1的范围的IR光谱示于图28。图的横轴为波数(cm-1),纵轴为吸光度(反射吸光度)。IR测定条件装置:FT-IR(BrukerOptics公司制)测定条件:ATR法(使用金刚石)测定环境:非活性气体环境下在图8表示的乙腈的IR光谱的2250cm-1附近,观察到来自乙腈的C和N间的三键的伸缩振动的特征峰。应予说明,在图9表示的(CF3SO2)2NLi的IR光谱和图10表示的(FSO2)2NLi的IR光谱的2250cm-1附近,没有观察到特别的峰。在图1表示的电解液E3的IR光谱中,在2250cm-1附近观察到微弱(Io=0.00699)的来自乙腈的C和N间的三键的伸缩振动的特征峰。此外在图1的IR光谱中,在从2250cm-1附近向高波数侧位移的2280cm-1附近以峰强度Is=0.05828观察到来自乙腈的C和N间的三键的伸缩振动的特征峰。Is与Io的峰强度的关系是Is>Io,Is=8×Io。在图2表示的电解液E4的IR光谱中,在2250cm-1附近没有观察到来自乙腈的峰,在从2250cm-1附近向高波数侧位移的2280cm-1附近以峰强度Is=0.05234观察到来自乙腈的C和N间的三键的伸缩振动的特征峰。Is与Io的峰强度的关系是Is>Io。在图3表示的电解液E7的IR光谱中,在2250cm-1附近观察到微弱(Io=0.00997)的来自乙腈的C和N间的三键的伸缩振动的特征峰。此外在图3的IR光谱中,在从2250cm-1附近向高波数侧位移的2280cm-1附近以峰强度Is=0.08288观察到来自乙腈的C和N间的三键的伸缩振动的特征峰。Is与Io的峰强度的关系是Is>Io,Is=8×Io。对于图4表示的电解液E8的IR光谱,也在同样的波数观察到与图3的IR图表同样的强度的峰。Is与Io的峰强度的关系是Is>Io,Is=11×Io。在图5表示的电解液E10的IR光谱中,在2250cm-1附近没有观察到来自乙腈的峰,在从2250cm-1附近向高波数侧位移的2280cm-1附近以峰强度Is=0.07350观察到来自乙腈的C和N间的三键的伸缩振动的特征峰。Is与Io的峰强度的关系是Is>Io。在图6表示的电解液C2的IR光谱中,与图8同样地,在2250cm-1附近以峰强度Io=0.04441观察到来自乙腈的C和N间的三键的伸缩振动的特征峰。此外在图6的IR光谱中,在从2250cm-1附近向高波数侧位移的2280cm-1附近以峰强度Is=0.03018观察到来自乙腈的C和N间的三键的伸缩振动的特征峰。Is与Io的峰强度的关系是Is<Io。在图7表示的电解液C4的IR光谱中,与图8同样地,在2250cm-1附近以峰强度Io=0.04975观察到来自乙腈的C和N间的三键的伸缩振动的特征峰。此外在图7的IR光谱中,在从2250cm-1附近向高波数侧位移的2280cm-1附近以峰强度Is=0.03804观察到来自乙腈的C和N间的三键的伸缩振动的特征峰。Is与Io的峰强度的关系是Is<Io。在图17表示的碳酸二甲酯的IR光谱的1750cm-1附近,观察到来自碳酸二甲酯的C和O间的双键的伸缩振动的特征峰。应予说明,在图28表示的(FSO2)2NLi的IR光谱的1750cm-1附近,没有观察到特别的峰。在图11表示的电解液E11的IR光谱中,在1750cm-1附近观察到微弱(Io=0.16628)的来自碳酸二甲酯的C和O间的双键的伸缩振动的特征峰。此外在图11的IR光谱中,在从1750cm-1附近向低波数侧位移的1717cm-1附近以峰强度Is=0.48032观察到来自碳酸二甲酯的C和O间的双键的伸缩振动的特征峰。Is与Io的峰强度的关系是Is>Io,Is=2.89×Io。在图12表示的电解液E12的IR光谱中,在1750cm-1附近观察到微弱(Io=0.18129)的来自碳酸二甲酯的C和O间的双键的伸缩振动的特征峰。此外在图12的IR光谱中,在从1750cm-1附近向低波数侧位移的1717cm-1附近以峰强度Is=0.52005观察到来自碳酸二甲酯的C和O间的双键的伸缩振动的特征峰。Is与Io的峰强度的关系是Is>Io,Is=2.87×Io。在图13表示的电解液E13的IR光谱中,在1750cm-1附近观察到微弱(Io=0.20293)的来自碳酸二甲酯的C和O间的双键的伸缩振动的特征峰。此外在图13的IR光谱中,在从1750cm-1附近向低波数侧位移的1717cm-1附近以峰强度Is=0.53091观察到来自碳酸二甲酯的C和O间的双键的伸缩振动的特征峰。Is与Io的峰强度的关系是Is>Io,Is=2.62×Io。在图14表示的电解液E14的IR光谱中,在1750cm-1附近观察到微弱(Io=0.23891)的来自碳酸二甲酯的C和O间的双键的伸缩振动的特征峰。此外在图14的IR光谱中,在从1750cm-1附近向低波数侧位移的1717cm-1附近以峰强度Is=0.53098观察到来自碳酸二甲酯的C和O间的双键的伸缩振动的特征峰。Is与Io的峰强度的关系是Is>Io,Is=2.22×Io。在图15表示的电解液E15的IR光谱中,在1750cm-1附近观察到微弱(Io=0.30514)的来自碳酸二甲酯的C和O间的双键的伸缩振动的特征峰。此外在图15的IR光谱中,在从1750cm-1附近向低波数侧位移的1717cm-1附近以峰强度Is=0.50223观察到来自碳酸二甲酯的C和O间的双键的伸缩振动的特征峰。Is与Io的峰强度的关系是Is>Io,Is=1.65×Io。在图16表示的电解液C6的IR光谱中,在1750cm-1附近观察到来自碳酸二甲酯的C和O间的双键的伸缩振动的特征峰(Io=0.48204)。此外在图16的IR光谱中,在从1750cm-1附近向低波数侧位移的1717cm-1附近以峰强度Is=0.39244观察到来自碳酸二甲酯的C和O间的双键的伸缩振动的特征峰。Is与Io的峰强度的关系是Is<Io。在图22表示的碳酸甲乙酯的IR光谱的1745cm-1附近,观察到来自碳酸甲乙酯的C和O间的双键的伸缩振动的特征峰。在图18表示的电解液E16的IR光谱中,在1745cm-1附近观察到微弱(Io=0.13582)的来自碳酸甲乙酯的C和O间的双键的伸缩振动的特征峰。此外在图18的IR光谱中,在从1745cm-1附近向低波数侧位移的1711cm-1附近以峰强度Is=0.45888观察到来自碳酸甲乙酯的C和O间的双键的伸缩振动的特征峰。Is与Io的峰强度的关系是Is>Io,Is=3.38×Io。在图19表示的电解液E17的IR光谱中,在1745cm-1附近观察到微弱(Io=0.15151)的来自碳酸甲乙酯的C和O间的双键的伸缩振动的特征峰。此外在图19的IR光谱中,在从1745cm-1附近向低波数侧位移的1711cm-1附近以峰强度Is=0.48779观察到来自碳酸甲乙酯的C和O间的双键的伸缩振动的特征峰。Is与Io的峰强度的关系是Is>Io,Is=3.22×Io。在图20表示的电解液E18的IR光谱中,在1745cm-1附近观察到微弱(Io=0.20191)的来自碳酸甲乙酯的C和O间的双键的伸缩振动的特征峰。此外在图20的IR光谱中,在从1745cm-1附近向低波数侧位移的1711cm-1附近以峰强度Is=0.48407观察到来自碳酸甲乙酯的C和O间的双键的伸缩振动的特征峰。Is与Io的峰强度的关系是Is>Io,Is=2.40×Io。在图21表示的电解液C7的IR光谱中,在1745cm-1附近观察到来自碳酸甲乙酯的C和O间的双键的伸缩振动的特征峰(Io=0.41907)。此外在图21的IR光谱中,在从1745cm-1附近向低波数侧位移的1711cm-1附近以峰强度Is=0.33929观察到来自碳酸甲乙酯的C和O间的双键的伸缩振动的特征峰。Is与Io的峰强度的关系是Is<Io。在图27表示的碳酸二乙酯的IR光谱的1742cm-1附近,观察到来自碳酸二乙酯的C和O间的双键的伸缩振动的特征峰。在图23表示的电解液E19的IR光谱中,在1742cm-1附近观察到微弱(Io=0.11202)的来自碳酸二乙酯的C和O间的双键的伸缩振动的特征峰。此外在图23的IR光谱中,在从1742cm-1附近向低波数侧位移的1706cm-1附近以峰强度Is=0.42925观察到来自碳酸二乙酯的C和O间的双键的伸缩振动的特征峰。Is与Io的峰强度的关系是Is>Io,Is=3.83×Io。在图24表示的电解液E20的IR光谱中,在1742cm-1附近观察到微弱(Io=0.15231)的来自碳酸二乙酯的C和O间的双键的伸缩振动的特征峰。此外在图24的IR光谱中,在从1742cm-1附近向低波数侧位移的1706cm-1附近以峰强度Is=0.45679观察到来自碳酸二乙酯的C和O间的双键的伸缩振动的特征峰。Is与Io的峰强度的关系是Is>Io,Is=3.00×Io。在图25表示的电解液E21的IR光谱中,在1742cm-1附近观察到微弱(Io=0.20337)的来自碳酸二乙酯的C和O间的双键的伸缩振动的特征峰。此外在图25的IR光谱中,在从1742cm-1附近向低波数侧位移的1706cm-1附近以峰强度Is=0.43841观察到来自碳酸二乙酯的C和O间的双键的伸缩振动的特征峰。Is与Io的峰强度的关系是Is>Io,Is=2.16×Io。在图26表示的电解液C8的IR光谱中,在1742cm-1附近观察到来自碳酸二乙酯的C和O间的双键的伸缩振动的特征峰(Io=0.39636)。此外在图26的IR光谱中,在从1742cm-1附近向低波数侧位移的1709cm-1附近以峰强度Is=0.31129观察到来自碳酸二乙酯的C和O间的双键的伸缩振动的特征峰。Is与Io的峰强度的关系是Is<Io。(评价例2:拉曼光谱测定)对电解液E8、E9、C4以及E11、E13、E15、C6,按以下的条件进行拉曼光谱测定。将观察到来自各电解液的金属盐的阴离子部分的峰的拉曼光谱分别示于图29~图35。图的横轴为波数(cm-1),纵轴为散射强度。拉曼光谱测定条件装置:激光拉曼光谱仪(日本分光株式会社NRS系列)激光波长:532nm在非活性气体环境下将电解液密闭于石英比色皿中,供于测定。在图29~图31表示的电解液E8、E9、C4的拉曼光谱的700~800cm-1,观察到来自溶解于乙腈的LiFSA的(FSO2)2N的特征峰。这里,由图29~图31可知随着LiFSA的浓度的增加,上述峰向高波数侧位移。随着电解液高浓度化,变成属于盐的阴离子的(FSO2)2N与Li相互作用的状态,换言之,推测浓度低时Li与阴离子主要形成了SSIP(Solvent-separatedionpairs:溶剂共享型离子对)状态,随着高浓度化,主要形成了CIP(Contactionpairs:接触型离子对)状态、AGG(aggregate:聚集)状态。而且,研究分析出该状态的变化以拉曼光谱的峰位移的形式被观察到。在图32~图35表示的电解液E11、E13、E15、C6的拉曼光谱的700~800cm-1,观察到来自溶解于碳酸二甲酯的LiFSA的(FSO2)2N的特征峰。这里,由图32~图35可知随着LiFSA的浓度的增加,上述峰向高波数侧位移。推测该现象与上一段分析的结果相同,是电解液高浓度化而使属于盐的阴离子的(FSO2)2N与多个Li相互作用的状态被反映到光谱的结果。(评价例3:离子传导率)按以下的条件测定电解液E1、E2、E4~E6、E8、E11、E16、E19的离子传导率。将结果示于表4。离子传导率测定条件在Ar环境下,将电解液封入具备铂极的电导池常数已知的玻璃制电导池中,以30℃、1kHz测定阻抗。由阻抗的测定结果计算离子传导率。测定设备使用Solartron147055BEC(Solartron公司)。表4电解液E1、E2、E4~E6、E8、E11、E16和E19均显示了离子传导性。因此,可以理解本发明的电解液均可作为各种电池的电解液发挥功能。(评价例4:粘度)按以下的条件测定电解液E1、E2、E4~6、E8、E11、E16、E19以及C1~C4、C6~C8的粘度。将结果示于表5。粘度测定条件使用落球式粘度计(AntonPaarGmbH(AntonPaar公司)制Lovis2000M),在Ar环境下,将电解液封入试验池,在30℃的条件下测定粘度。表5电解液E1、E2、E4~6、E8、E11、E16、E19的粘度与电解液C1~C4,C6~C8的粘度相比较,明显高。因此,如果为使用了本发明的电解液的电池,则即便电池破损,也能抑制电解液泄漏。(评价例5:挥发性)用以下的方法测定电解液E2、E4、E8、E11、E13、C1、C2、C4和C6的挥发性。将约10mg的电解液放入铝制的锅内,配置于热重测定装置(TAInstruments公司制,SDT600),测定室温下的电解液的重量变化。将重量变化(质量%)用时间进行微分而计算挥发速度。选择挥发速度中最大的速度,示于表6。表6电解液E2、E4、E8、E11、E13的最大挥发速度与电解液C1、C2、C4、C6的最大挥发速度相比较,明显小。因此,使用了本发明的电解液的电池即便发生损伤,电解液的挥发速度也小,所以抑制了有机溶剂向电池外的快速挥发。(评价例6:燃烧性)用以下的方法对电解液E4、C2的燃烧性进行试验。用滴管将电解液向玻璃滤材(glassfilters)滴加3滴,使电解液保持在玻璃滤材。用镊子夹持该玻璃滤材,然后,使该玻璃滤材接触火焰。电解液E4与火焰接触15秒也没有起火。另一方面,电解液C2经过5秒多就燃尽了。证明了本发明的电解液不易燃烧。以下,对非水电解质二次电池(1)和非水电解质二次电池(2)进行具体说明。以下的实施例和EB、CB,为了方便,分项说明,因此有时会重复。另外,以下的实施例和后述的EB、CB有时属于非水电解质二次电池(1)和非水电解质二次电池(2)这两者的实施例。(EB1)如下制造使用了电解液E8的半电池。将作为活性物质的平均粒径10μm的石墨90质量份和作为粘结剂的聚偏氟乙烯10质量份混合。使该混合物分散于适量的N-甲基-2-吡咯烷酮,制作浆料。准备厚度20μm的铜箔作为集电体。使用刮刀,在该铜箔的表面将上述浆料涂布成膜状。将涂布有浆料的铜箔干燥而除去N-甲基-2-吡咯烷酮,其后,对铜箔加压,得到接合物。将得到的接合物用真空干燥机在120℃加热干燥6小时,得到形成有活性物质层的铜箔。将其作为工作电极。对电极为金属Li。将工作电极、对电极、夹在两者之间的作为隔离件的厚度400μm的Whatman玻璃纤维滤纸和电解液E8收容在电池盒(宝泉株式会社制CR2032型纽扣电池盒)中,得到非水电解质二次电池EB1。该非水电解质二次电池是评价用的非水电解质二次电池,也称为半电池。(CB1)使用电解液C5,除此之外,用与EB1同样的方法,制造非水电解质二次电池CB1。(评价例7:倍率特性)用以下的方法对EB1、CB1的倍率特性进行试验。对于各非水电解质二次电池,以0.1C、0.2C、0.5C、1C、2C倍率(1C是指在一定电流下经1小时使电池完全充电或放电所需的电流值)进行充电后进行放电,测定各速度下的工作电极的容量(放电容量)。应予说明,这里的记述是将对电极看作负极,将工作电极看作正极。计算其它倍率下的容量相对于0.1C倍率下的工作电极的容量的比例(倍率特性)。将结果示于表7。表7EB1CB10.1C容量(mAh/g)3343300.2C容量/0.1C容量0.9830.9660.5C容量/0.1C容量0.9460.7671C容量/0.1C容量0.8680.4982C容量/0.1C容量0.4710.177在0.2C、0.5C、1C、2C的任一倍率下,EB1与CB1相比较,均抑制了容量的降低,显示了优异的倍率特性。证明了使用本发明的电解液的二次电池显示优异的倍率特性。(评价例8:相对于反复快速充放电的响应性)观察以1C倍率对非水电解质二次电池EB1和CB1反复充放电3次时的容量和电压的变化。将结果示于图36。CB1随着反复充放电,有以1C倍率流动电流时的极化变大的趋势,从2V到0.01V得到的容量快速降低。另一方面,EB1即便经过反复充放电,从图36中3条曲线重叠的样子也能够确认极化几乎没有增减,良好地维持了容量。作为CB1中极化增加的理由,认为是由于快速反复充放电时的电解液中产生的Li浓度不均,导致电解液无法向与电极的反应界面供给足够量的Li,也就是说,电解液的Li浓度不均。认为EB1中,通过使用Li浓度高的本发明的电解液,能够抑制电解液的Li浓度的不均。证明了使用本发明的电解液的二次电池对于快速充放电显示优异的响应性。(评价例9:Li迁移数)按以下的条件测定电解液E2、E8、C4和C5的Li迁移数。将结果示于表8。(Li迁移数测定条件)将装有电解液的NMR管供给于PFG-NMR装置(ECA-500,日本电子),以7Li、19F为对象,利用自旋回波法,边使磁场脉冲宽度变化,边测定各电解液中的Li离子和阴离子的扩散系数。Li迁移数用以下的式子计算。Li迁移数=(Li离子扩散系数)/(Li离子扩散系数+阴离子扩散系数)表8电解液E2、E8的Li迁移数与电解液C4、C5的Li迁移数相比明显高。这里,电解液的Li离子传导率可以通过使电解液所含的离子传导率(总离子电导率)乘以Li迁移数而计算。于是,可以说本发明的电解液与显示同等程度的离子传导率的现有电解液相比,锂离子(阳离子)的输送速度高。另外,对于电解液E8,基于上述Li迁移数测定条件测定温度变化时的Li迁移数。将结果示于表9。表9温度(℃)Li迁移数300.50100.50-100.50-300.52由表9的结果可知本发明的电解液不依赖于温度,保持良好的Li迁移数。可以说本发明的电解液在低温下也保持了液体状态。〔非水电解质二次电池〕(EB2)如下制造使用了电解液E8的非水电解质二次电池EB2。将作为正极活性物质的由LiNi5/10Co2/10Mn3/10O2表示的层状岩盐结构的含锂金属氧化物94质量份、作为导电助剂的乙炔黑3质量份和作为粘结剂的聚偏氟乙烯3质量份混合。使该混合物分散于适量的N-甲基-2-吡咯烷酮,制作浆料。准备厚度20μm的铝箔(JISA1000号)作为正极集电体。使用刮刀,将上述浆料以成为膜状的方式涂布在该铝箔的表面。将涂布有浆料的铝箔在80℃干燥20分钟通过挥发而除去N-甲基-2-吡咯烷酮。其后,对该铝箔加压得到接合物。将得到的接合物用真空干燥机在120℃加热干燥6小时,得到形成有正极活性物质层的铝箔。将其作为正极。以下,根据需要,将由LiNi5/10Co2/10Mn3/10O2表示的层状岩盐结构的含锂金属氧化物省略为NCM523,将乙炔黑省略为AB,将聚偏氟乙烯省略为PVdF。将作为负极活性物质的天然石墨98质量份以及作为粘结剂的苯乙烯丁二烯橡胶1质量份和羧甲基纤维素1质量份混合。使该混合物分散于适量的离子交换水,制作浆料。准备厚度20μm的铜箔作为负极集电体。使用刮刀,在该铜箔的表面将上述浆料涂布成膜状。将涂布有浆料的铜箔干燥而除去水,其后,对铜箔加压,得到接合物。将得到的接合物用真空干燥机在100℃加热干燥6小时,得到形成有负极活性物质层的铜箔。将其作为负极。以下,根据需要,将苯乙烯丁二烯橡胶省略为SBR,将羧甲基纤维素省略为CMC。作为隔离件,准备厚度20μm的纤维素制无纺布。用正极和负极夹持隔离件,制成极板组。将该极板组用二片一组的层压膜覆盖,将三边进行密封后,向呈袋状的层压膜注入电解液E8。其后,将剩余的一边进行密封,由此得到四边被气密地密封、极板组和电解液被密闭的非水电解质二次电池。将该电池作为非水电解质二次电池EB2。(EB3)如下制造使用了电解液E8的非水电解质二次电池EB3。正极与非水电解质二次电池EB2的正极同样地进行制造。将作为负极活性物质的天然石墨90质量份和作为粘结剂的聚偏氟乙烯10质量份混合。使该混合物分散于适量的离子交换水,制作浆料。准备厚度20μm的铜箔作为负极集电体。使用刮刀,在该铜箔的表面将上述浆料涂布成膜状。将涂布有浆料的铜箔干燥而除去水,其后,对铜箔加压,得到接合物。将得到的接合物用真空干燥机在120℃加热干燥6小时,得到形成有负极活性物质层的铜箔。将其作为负极。作为隔离件,准备厚度20μm的纤维素制无纺布。用正极和负极夹持隔离件,制成极板组。将该极板组用二片一组的层压膜覆盖,将三边进行密封后,向呈袋状的层压膜注入电解液E8。其后,将剩余的一边进行密封,由此得到层压膜的四边被密封、极板组和电解液被密闭在该层压膜内的非水电解质二次电池。将该电池作为非水电解质二次电池EB3。(CB2)使用电解液C5,除此之外,与EB2同样地制造非水电解质二次电池CB2。(CB3)使用电解液C5,除此之外,与EB3同样地制造非水电解质二次电池CB3。(评价例10:非水电解质二次电池的输入输出特性)按以下的条件评价非水电解质二次电池EB2、EB3、CB2、CB3的输出特性。(1)0℃或25℃、SOC80%时的输入特性评价评价条件设为充电状态(SOC)80%、0℃或25℃、使用电压范围3V―4.2V、容量13.5mAh。输入特性的评价是对每个电池分别进行3次2秒输入和5秒输入。另外,基于各电池的体积,计算25℃、2秒输入时的电池输出密度(W/L)。将输入特性的评价结果示于表10。表10中的“2秒输入”是指在充电开始2秒后的输入,“5秒输入”是指在充电开始5秒后的输入。如表10所示,与温度的差异无关,EB2的输入与CB2的输入相比,明显高。同样,EB3的输入与CB3的输入相比,明显高。另外,EB2的电池输入密度与CB2的电池输入密度相比,明显高。同样,EB3的电池输入密度与CB3的电池输入密度相比,明显高。(2)0℃或25℃、SOC20%时的输出特性评价评价条件设为充电状态(SOC)20%、0℃或25℃、使用电压范围3V―4.2V、容量13.5mAh。SOC20%、0℃例如是像在冷藏室等使用时那样输出特性不易体现的区域。输出特性的评价是对各电池分别进行3次2秒输出和5秒输出。另外,基于各电池的体积,计算25℃、2秒输出时的电池输出密度(W/L)。将输出特性的评价结果示于表10。表10中的“2秒输出”是指在放电开始2秒后的输出,“5秒输出”是指在放电开始5秒后的输出。如表10所示,与温度的差异无关,EB2的输出与CB2的输出相比明显高。同样,EB3的输出与CB3的输出相比,明显高。另外,EB2的电池输出密度与CB2的电池输出密度相比明显高。同样,EB3的电池输出密度与CB3的电池输出密度相比明显高。表10(评价例11:低温试验)将电解液E11、E13、E16、E19分别放入容器,填充非活性气体进行密闭。将它们在-30℃的冰箱保管2天。保管后观察各电解液。任何电解液均没有固化而维持了液体状态,也没有观察到盐的析出。(实施例1-1)如下制造使用了电解液E8的实施例1-1的非水电解质二次电池。正极与非水电解质二次电池EB2的正极同样地进行制造。将作为负极活性物质的天然石墨98质量份以及作为粘结剂的SBR1质量份和CMC1质量份混合。使该混合物分散于适量的离子交换水,制作浆料。准备厚度20μm的铜箔作为负极集电体。使用刮刀,在该铜箔的表面将上述浆料涂布成膜状。将涂布有浆料的铜箔干燥而除去水,其后,对铜箔加压,得到接合物。将得到的接合物用真空干燥机在100℃加热干燥6小时,得到形成有负极活性物质层的铜箔。将其作为负极。作为隔离件,准备实验用滤纸(东洋滤纸株式会社,纤维素制,厚度260μm)。用正极和负极夹持隔离件,制成极板组。将该极板组用二片一组的层压膜覆盖,将三边进行密封后,向呈袋状的层压膜注入电解液E8。其后,将剩余的一边进行密封,由此得到四边被气密地密封、极板组和电解液被密闭的非水电解质二次电池。将该电池作为实施例1-1的非水电解质二次电池。(实施例1-2)实施例1-2的非水电解质二次电池除使用电解液E4作为电解液以外,与实施例1-1的非水电解质二次电池相同。实施例1-2的非水电解质二次电池中的电解液是在作为溶剂的乙腈中溶解作为支持盐的(SO2CF3)2NLi(LiTFSA)而成的。1升电解液所含的锂盐的浓度为4.2mol/L。电解液中,相对于锂盐1分子,含有2分子的乙腈。(实施例1-3)实施例1-3的非水电解质二次电池除使用电解液E11作为电解液以外,与实施例1-1的非水电解质二次电池相同。实施例1-3的非水电解质二次电池中的电解液是在作为溶剂的DMC中溶解作为支持盐的LiFSA而成的。1升电解液所含的锂盐的浓度为3.9mol/L。电解液中,相对于锂盐1分子,含有2分子的DMC。(实施例1-4)实施例1-4的非水电解质二次电池使用了电解液E11。实施例1-4的非水电解质二次电池除电解液的种类、正极活性物质与导电助剂与粘结剂的混合比、负极活性物质与粘结剂的混合比和隔离件以外,与实施例1-1的非水电解质二次电池相同。对于正极,使用NCM523作为正极活性物质,使用AB作为正极用的导电助剂,使用PVdF作为粘结剂。其与实施例1-1是同样的。它们的配合比是NCM523:AB:PVdF=90:8:2。正极中的活性物质层的单位面积重量为5.5mg/cm2,密度为2.5g/cm3。其对于以下的实施例1-5~1-7和比较例1-2、1-3也是同样的。对于负极,使用天然石墨作为负极活性物质,使用SBR和CMC作为负极用的粘结材料。另外其也与实施例1-1是同样的。它们的配合比是天然石墨:SBR:CMC=98:1:1。负极中的活性物质层的单位面积重量为3.8mg/cm2,密度为1.1g/cm3。其对于以下的实施例1-5~1-7和比较例1-2、1-3也是同样的。作为隔离件,使用了厚度20μm的纤维素制无纺布。实施例1-4的非水电解质二次电池中的电解液是在作为溶剂的DMC中溶解作为支持盐的LiFSA而成的。1升电解液所含的锂盐的浓度为3.9mol/L。电解液中,相对于锂盐1分子,含有2分子的DMC。(实施例1-5)实施例1-5的非水电解质二次电池使用了电解液E8。实施例1-5的非水电解质二次电池除正极活性物质与导电助剂与粘结剂的混合比、负极活性物质与粘结剂的混合比以及隔离件以外,与实施例1-1的非水电解质二次电池相同。对于正极,是NCM523:AB:PVdF=90:8:2。对于负极,是天然石墨:SBR:CMC=98:1:1。作为隔离件,使用了厚度20μm的纤维素制无纺布。(实施例1-6)实施例1-6的非水电解质二次电池使用了电解液E11。实施例1-6的非水电解质二次电池除电解液的种类、正极活性物质与导电助剂与粘结剂的混合比、负极用的粘结材料的种类、负极活性物质与粘结剂的混合比和隔离件以外,与实施例1-1的非水电解质二次电池相同。对于正极,是NCM523:AB:PVdF=90:8:2。对于负极,使用天然石墨作为负极活性物质,使用聚丙烯酸(PAA)作为负极用的粘结材料。它们的配合比是天然石墨:PAA=90:10。作为隔离件,使用了厚度20μm的纤维素制无纺布。(实施例1-7)实施例1-7的非水电解质二次电池使用了电解液E8。实施例1-7的非水电解质二次电池除正极活性物质与导电助剂与粘结剂的混合比、负极用的粘结材料的种类、负极活性物质与粘结剂的混合比和隔离件以外,与实施例1-1的非水电解质二次电池相同。对于正极,是NCM523:AB:PVdF=90:8:2。对于负极,是天然石墨:PAA=90:10。作为隔离件,使用了厚度20μm的纤维素制无纺布。(实施例1-8)实施例1-8的非水电解质二次电池使用了电解液E13。实施例1-8的非水电解质二次电池除正极活性物质与导电助剂的混合比、负极用的粘结材料的种类、负极活性物质与粘结剂的混合比和隔离件以外,与实施例1-1的非水电解质二次电池相同。对于正极,是NCM523:AB:PVdF=90:8:2。对于负极,是天然石墨:SBR:CMC=98:1:1。作为隔离件,使用了厚度20μm的纤维素制无纺布。(比较例1-1)比较例1-1的非水电解质二次电池使用电解液C5作为电解液,除此之外,与实施例1-1同样。(比较例1-2)比较例1-2的非水电解质二次电池使用了电解液C5。比较例1-2的非水电解质二次电池除电解液的种类、正极活性物质与导电助剂与粘结剂的混合比、负极活性物质与粘结剂的混合比和隔离件以外,与实施例1-1的非水电解质二次电池相同。对于正极,是NCM523:AB:PVdF=90:8:2。对于负极,是天然石墨:SBR:CMC=98:1:1。作为隔离件,使用了厚度20μm的纤维素制无纺布。(比较例1-3)比较例1-3的非水电解质二次电池使用了电解液C5。比较例1-3的非水电解质二次电池除电解液的种类、正极活性物质与导电助剂与粘结剂的混合比、负极用的粘结材料的种类、负极活性物质与粘结剂的混合比和隔离件以外,与实施例1-1的非水电解质二次电池相同。对于正极,是NCM523:AB:PVdF=90:8:2。对于负极,是天然石墨:PAA=90:10。作为隔离件,使用了厚度20μm的纤维素制无纺布。将实施例和比较例的电池构成示于表11。表11(评价例12:含S、O被膜的分析)以下,根据需要,将在各实施例的非水电解质二次电池中的负极的表面形成的含S、O被膜简称为各实施例的负极含S、O被膜,将在各比较例的非水电解质二次电池中的负极的表面形成的被膜简称为各比较例的负极被膜。另外,根据需要,将在各实施例的非水电解质二次电池中的正极的表面形成的被膜简称为各实施例的正极含S、O被膜,将在各比较例的非水电解质二次电池中的正极的表面形成的被膜简称为各比较例的正极被膜。(负极含S、O被膜和负极被膜的分析)对于实施例1-1、实施例1-2和比较例1-1的非水电解质二次电池反复进行100次循环充放电后,在电压3.0V的放电状态下通过X射线光电子能谱(X-rayPhotoelectronSpectroscopy,XPS)进行含S、O被膜或被膜表面的分析。作为前处理,进行以下的处理。首先,将非水电解质二次电池解体取出负极,并将该负极进行清洗和干燥,得到作为分析对象的负极。清洗使用DMC(碳酸二甲酯)进行3次。另外,从电池的解体到将作为分析对象的负极输送到分析装置的全部工序是在Ar气环境下不使负极与大气接触而进行的。对实施例1-1、实施例1-2和比较例1-1的各非水电解质二次电池进行以下的前处理,将得到的负极检体进行XPS分析。作为装置,使用ULVAC-PHI公司PHI5000VersaProbeII。X射线源为单色AlKα射线(15kV,10mA)。将利用XPS测定的实施例1-1、实施例1-2的负极含S、O被膜和比较例1-1的负极被膜的分析结果示于图37~图41。具体而言,图37是针对碳元素的分析结果,图38是针对氟元素的分析结果,图39是针对氮元素的分析结果,图40是针对氧元素的分析结果,图41是针对硫元素的分析结果。实施例1-1的非水电解质二次电池的电解液和实施例1-2的非水电解质二次电池的电解液的盐中含有硫元素(S)、氧元素和氮元素(N)。与此相对,比较例1-1的非水电解质二次电池的电解液的盐中不含这些元素。此外,实施例1-1、实施例1-2和比较例1-1的非水电解质二次电池的电解液均在盐中含有氟元素(F)、碳元素(C)和氧元素(O)。如图37~图41所示,对实施例1-1的负极含S、O被膜和实施例1-2的负极含S、O被膜进行分析,结果观察到显示S的存在的峰(图41)和显示N的存在的峰(图39)。也就是说,实施例1-1的负极含S、O被膜和实施例1-2的负极含S、O被膜含有S和N。但是,在比较例1-1的负极被膜的分析结果中没有发现这些峰。也就是说,比较例1-1的负极被膜不含有检出限以上的量的S和N中的任一个。应予说明,显示F、C和O的存在的峰在实施例1-1、实施例1-2的负极含S、O被膜和比较例1-1的负极被膜的全部分析结果中被观察到。也就是说,实施例1-1、实施例1-2的负极含S、O被膜和比较例1-1的负极被膜均含有F、C和O。这些元素均是来自电解液的成分。特别是S、O和F是电解液的金属盐所含的成分,具体而言是金属盐的阴离子的化学结构所含的成分。因此,由这些结果可知各负极含S、O被膜和负极被膜含有来自于金属盐(也就是说支持盐)的阴离子的化学结构的成分。对图41所示的硫元素(S)的分析结果进行更详细地解析。针对实施例1-1和实施例1-2的分析结果,利用高斯/洛伦兹混合函数进行峰分离。将实施例1-1的解析结果示于图42,将实施例1-2的解析结果示于图43。如图42和图43所示,对实施例1-1和1-2的负极含S、O被膜进行分析,结果在165~175eV附近观察到较大的峰(波形)。而且,如图42和图43所示,该170eV附近的峰(波形)被分离成4个峰。其中一个是表示SO2(S=O结构)的存在的170eV附近的峰。根据该结果,可以说在本发明的非水电解质二次电池中负极表面形成的含S、O被膜具有S=O结构。而且,考虑该结果和上述的XPS分析结果,推测含S、O被膜的S=O结构所含的S是金属盐即支持盐的阴离子的化学结构所含的S。(负极含S、O被膜的S元素比率)基于上述的负极含S、O被膜的XPS分析结果,计算实施例1-1和实施例1-2的负极含S、O被膜和比较例1-1的负极被膜中的放电时的S元素的比率。具体而言,对各负极含S、O被膜和负极被膜,计算将S、N、F、C、O的峰强度的总和设为100%时的S的元素比。将结果示于表12。表12实施例1-1实施例1-2比较例1-1S元素比率(原子%)10.43.70.0如上所述,比较例1-1的负极被膜不含检出限以上的S,但从实施例1-1的负极含S、O被膜和实施例1-2的负极含S、O被膜检测到S。另外,实施例1-1的负极含S、O被膜与实施例1-2的负极含S、O被膜相比,含有更多的S。应予说明,从比较例1-1的负极含S、O被膜没有检测到S,因此可以说各实施例的负极含S、O被膜所含的S并非来自正极活性物质所含的不可避免的杂质或其它的添加物,而是来自电解液中的金属盐。另外,实施例1-1的负极含S、O被膜中的S元素比率为10.4原子%,实施例1-2的负极含S、O被膜中的S元素比率为3.7原子%,因此,在本发明的非水电解质二次电池中,负极含S、O被膜中的S元素比率为2.0原子%以上,优选为2.5原子%以上,更优选为3.0原子%以上,进一步优选为3.5原子%以上。应予说明,如上所述S的元素比率(原子%)是指将S、N、F、C、O的峰强度的总和设为100%时的S的峰强度比。S的元素比率的上限值没有特别限定,非要要求的话,优选为25原子%以下。(负极含S、O被膜的厚度)准备对实施例1-1的非水电解质二次电池反复进行100次循环充放电后成为电压3.0V的放电状态的电池和反复进行100次循环充放电后成为电压4.1V的充电状态的电池,用与上述的XPS分析的前处理同样的方法得到作为分析对象的负极检体。通过对得到的负极检体进行FIB(聚焦离子束:FocusedIonBeam)加工,得到厚度100nm左右的STEM分析用检体。应予说明,作为FIB加工的前处理,在负极上蒸镀了Pt。以上的工序在不使负极与大气接触的条件下进行。利用附带有EDX(能量色散型X射线谱:EnergyDispersiveX-rayspectroscopy)装置的STEM(扫描透射电子显微镜:ScanningTransmissionElectronMicroscope)分析各STEM分析用检体。将结果示于图44~图47。其中图44是BF(明视野:Bright-field)-STEM图像,图45~图47是与图44相同的观察区域的利用SETM-EDX得到的元素分布图像。此外,图45是针对C的分析结果,图46是针对O的分析结果,图47是针对S的分析结果。应予说明,图45~图47是放电状态的非水电解质二次电池中的负极的分析结果。如图44所示,在STEM图像的左上部存在黑色的部分。该黑色的部分来自在FIB加工的前处理中被蒸镀的Pt。各STEM图像中,在该来自Pt的部分(称为Pt部)的上侧的部分可看作是Pt蒸镀后被污染的部分。因此,图45~图47中,仅对在Pt部的下侧的部分进行研究。如图45所示,在Pt部的下侧,C呈现层状。认为这是作为负极活性物质的石墨的层状结构。图46中,O位于与石墨的外周和层间相当的部分。另外,图47中S也位于与石墨的外周和层间相当的部分。由这些结果推测含有S=O结构等的S和O的负极含S、O被膜在石墨的表面和层间形成。随机选择10处在石墨的表面形成的负极含S、O被膜,测定负极含S、O被膜的厚度,计算测定值的平均值。对充电状态的非水电解质二次电池中的负极也同样地分析,基于各分析结果,计算在石墨的表面形成的负极含S、O被膜的厚度的平均值。将结果示于表13。表13如表13所示,负极含S、O被膜的厚度在充电后增加。由该结果推测负极含S、O被膜中存在相对于充放电稳定存在的固定部和伴随充放电增减的吸附部。而且,推测由于吸附部存在,负极含S、O被膜在充放电时厚度发生增减。(正极被膜的分析)准备对实施例1-1的非水电解质二次电池反复进行3次循环充放电后成为电压3.0V的放电状态的电池、反复进行3次循环充放电后成为电压4.1V的充电状态的电池、反复进行100次循环充放电后成为电压3.0V的放电状态的电池、反复进行100次循环充放电后成为电压4.1V的充电状态的电池共4个。对这4个实施例1-1的非水电解质二次电池分别使用与上述同样的方法,得到作为分析对象的正极。然后对得到的各正极进行XPS分析。将结果示于图48和图49。应予说明,图48是针对氧元素的分析结果,图49是针对硫元素的分析结果。如图48和图49所示,可知实施例1-1的正极含S、O被膜也另外含有S和O。另外,由于在图49中出现170eV附近的峰,所以可知实施例1-1的正极含S、O被膜与实施例1-1的负极含S、O被膜同样地也另外具有来自本发明的电解液的S=O结构。然而,如图48所示,在529eV附近存在的峰的高度经过循环后减少。认为该峰表示来自正极活性物质的O的存在,具体而言,XPS分析中被正极活性物质中的O原子激发的光电子通过含S、O被膜被检测到。由于该峰经过循环后减少,所以认为在正极表面形成的含S、O被膜的厚度随着循环的进行而增大。另外,如图48和图49所示,正极含S、O被膜中的O和S在放电时增加,充电时减少。根据该结果,认为O和S伴随着充放电出入正极含S、O被膜。而且由此推测出在充放电时正极含S、O被膜中的S、O的浓度发生增减,或者与负极含S、O被膜同样地正极含S、O被膜中也由于吸附部的存在而使厚度发生增减。此外,对于实施例1-4的非水电解质二次电池,也将正极含S、O被膜和负极含S、O被膜进行XPS分析。使实施例1-4的非水电解质二次电池成为25℃、使用电压范围3.0V~4.1V,以倍率1C反复500次循环CC充放电。500次循环后,在3.0V的放电状态和4.0V的充电状态下测定正极含S、O被膜的XPS光谱。另外,对循环试验前(也就是说初次充放电后)的3.0V的放电状态的负极含S、O被膜和500循环后的3.0V的放电状态的负极含S、O被膜进行基于XPS的元素分析,计算该负极含S、O被膜所含的S元素比率。将利用XPS测定的实施例1-4的正极含S、O被膜的分析结果示于图50和图51。具体而言,图50是针对硫元素的分析结果,图51是针对氧元素的分析结果。另外,将利用XPS测定的负极被膜的S元素比率(原子%)示于表14。应予说明,S元素比率与上述的“负极含S、O被膜的S元素比率”一项同样地计算。如图50和图51所示,从实施例1-4的非水电解质二次电池中的正极含S、O被膜也另外检测到表示S的存在的峰和表示O的存在的峰。另外,S的峰和O的峰均在放电时增大,充电时减少。根据该结果,证明了正极含S、O被膜具有S=O结构,正极含S、O被膜中的O和S伴随充放电出入正极含S、O被膜。表14<负极含S、O被膜的S元素比率>初次充放电后500次循环后S元素比率(原子%)3.13.8另外,如表14所示,实施例1-4的负极含S、O被膜在初次充放电后、在经过500次循环后,均含有2.0原子%以上的S。由该结果可知本发明的非水电解质二次电池中的负极含S、O被膜在经过循环前和经过循环后均含有2.0原子%以上的S。对实施例1-4~实施例1-7和比较例1-2、比较例1-3的非水电解质二次电池进行在60℃储藏1周的高温储藏试验,对该高温储藏试验后的各实施例的正极含S、O被膜和负极含S、O被膜以及各比较例的正极被膜和负极被膜进行分析。在高温储藏试验开始前,以倍率0.33C从3.0V进行CC-CV充电至4.1V。以此时的充电容量为基准(SOC100),相对于该基准CC放电20%部份而调整成SOC80后,开始高温储藏试验。在高温储藏试验后以1C进行CC-CV放电至3.0V。然后,测定放电后的正极含S、O被膜和负极含S、O被膜以及正极被膜和负极被膜的XPS光谱。将利用XPS测定的实施例1-4~实施例1-7的正极含S、O被膜以及比较例1-2和比较例1-3的正极被膜的分析结果示于图52~图55。另外,将利用XPS测定的实施例1-4~实施例1-7的负极含S、O被膜以及比较例1-2和比较例1-3的负极被膜的分析结果示于图56~图52。具体而言,图52是针对实施例1-4、实施例1-5的正极含S、O被膜和比较例1-2的正极被膜的硫元素的分析结果。图53是针对实施例1-6、实施例1-7的正极含S、O被膜和比较例1-3的正极被膜的硫元素的分析结果。图54是针对实施例1-4、实施例1-5的正极含S、O被膜和比较例1-2的正极被膜的氧元素的分析结果。图55是针对实施例1-6、实施例1-7的正极含S、O被膜和比较例1-3的正极被膜的氧元素的分析结果。另外,图56是针对实施例1-4、实施例1-5的负极含S、O被膜和比较例1-2的负极被膜的硫元素的分析结果。图57是针对实施例1-6、实施例1-7的负极含S、O被膜和比较例1-3的负极被膜的硫元素的分析结果。图58是针对实施例1-4、实施例1-5的负极含S、O被膜和比较例1-2的负极被膜的氧元素的分析结果。图59是针对实施例1-6、实施例1-7的负极含S、O被膜和比较例1-3的负极被膜的氧元素的分析结果。如图52和图53所示,使用了现有的电解液的比较例1-2和比较例1-3的非水电解质二次电池的正极被膜中不含S,与此相对,使用了本发明的电解液的实施例1-4~实施例1-7的非水电解质二次电池的正极含S、O被膜中含有S。另外,如图54和图55所示,实施例1-4~实施例1-7的非水电解质二次电池的正极含S、O被膜中均含有O。此外,如图52和图53所示,从实施例1-4~实施例1-7的非水电解质二次电池中的正极含S、O被膜均检测到表示SO2(S=O结构)的存在的170eV附近的峰。由这些结果可知在本发明的非水电解质二次电池中,使用AN、DMC作为电解液用的有机溶剂时,均形成含有S和O的稳定的正极含S、O被膜。另外,由于该正极含S、O被膜不受负极粘结剂的种类影响,所以认为正极含S、O被膜中的O并非来自CMC。此外,如图54和图55所示,使用DMC作为电解液用的有机溶剂时,在530eV附近检测到来自正极活性物质的O峰。因此,认为使用DMC作为电解液用的有机溶剂时,与使用AN的情况相比,正极含S、O被膜的厚度变薄。同样地,由图56~图59可知实施例1-4~实施例1-7的非水电解质二次电池的负极含S、O被膜也含有S和O,它们形成S=O结构且来自电解液。而且可知在使用AN、DMC作为电解液用的有机溶剂时均形成该负极含S、O被膜。对实施例1-4、实施例1-5和比较例1-2的非水电解质二次电池测定上述的高温储藏试验和放电后的各负极含S、O被膜以及负极被膜的XPS光谱,计算实施例1-4、实施例1-5的负极含S、O被膜和比较例1-2的负极被膜中的放电时的S元素的比率。具体而言,对各负极含S、O被膜或负极被膜计算将S、N、F、C、O的峰强度的总和设为100%时的S的元素比。将结果示于表15。表15实施例1-4实施例1-5比较例1-2S元素比率(原子%)4.26.40.0如表15所示,比较例1-2的负极被膜不含检出限以上的S,但从实施例1-4和实施例1-5的负极含S、O被膜检测出S。另外,实施例1-5的负极含S、O被膜与实施例1-4的负极含S、O被膜相比,含有更多的S。另外,由该结果可知即便在高温储藏后负极含S、O被膜中的S元素比率也为2.0原子%以上。(评价例13:电池的内部电阻)准备实施例1-4、实施例1-5、实施例1-8和比较例1-2的非水电解质二次电池,评价电池的内部电阻。对实施例1-4、实施例1-5、实施例1-8和比较例1-2的各非水电解质二次电池,在室温、3.0V~4.1V(vs.Li基准)的范围反复进行CC充放电(也就是说恒定电流充放电)。然后,测定初次充放电后的交流阻抗和经过100次循环后的交流阻抗。基于得到的复阻抗平面曲线,分别解析电解液、负极和正极的反应电阻。如图60所示,在复阻抗平面曲线中,看到二个圆弧。将图中左侧(即复阻抗的实部小的一侧)的圆弧称为第1圆弧。将图中右侧的圆弧称为第2圆弧。根据第1圆弧的大小解析负极的反应电阻,根据第2圆弧的大小解析正极的反应电阻。根据与第1圆弧连接的图60中最左侧的曲线解析电解液的电阻。将解析结果示于表16和表17。应予说明,表16示出初次充放电后的电解液的电阻(所谓溶液电阻)、负极的反应电阻、正极的反应电阻,表17示出经过100次循环后的各电阻。表16〈初始交流电阻〉单位:Ω表17<100次循环后的交流电阻〉单位:Ω如表16和表17所示,各非水电解质二次电池中,经过100次循环后的负极反应电阻和正极反应电阻与初次充放电后的各电阻相比有降低的趋势。而且,在经过表17所示的100次循环后,各实施例的非水电解质二次电池的负极反应电阻和正极反应电阻比比较例1-2的非水电解质二次电池的负极反应电阻和正极反应电阻低。如上所述,实施例1-4、实施例1-5和实施例1-8的非水电解质二次电池使用了本发明的电解液,在负极和正极的表面形成了来自本发明的电解液的含S、O被膜。与此相对,在没有使用本发明的电解液的比较例1-2的非水电解质二次电池中,在负极和正极的表面没有形成该含S、O被膜。而且,如表17所示,实施例1-4、实施例1-5和实施例1-8的负极反应电阻和正极反应电阻比比较例1-2的非水电解质二次电池低。由此,推测各实施例中,由于来自本发明的电解液的含S、O被膜的存在,使负极反应电阻和正极反应电阻减少。应予说明,实施例1-5和比较例1-2的非水电解质二次电池中的电解液的溶液电阻几乎相同,实施例1-4和实施例1-8的非水电解质二次电池中的电解液的溶液电阻比实施例1-5和比较例1-2高。另外,各非水电解质二次电池中的各电解液的溶液电阻在初次充放电后和经过100次循环后也几乎相同。因此,认为没有发生各电解液的耐久劣化,认为上述的比较例和实施例中产生的负极反应电阻和正极反应电阻的差与电解液的耐久劣化无关,而是由电极本身产生的。非水电解质二次电池的内部电阻可以由电解液的溶液电阻、负极的反应电阻和正极的反应电阻综合判断。基于表16和表17的结果,从抑制非水电解质二次电池的内部电阻增大的观点考虑,可以说实施例1-4和实施例1-8的非水电解质二次电池的耐久性特别优异,接着实施例1-5的非水电解质二次电池的耐久性优异。(评价例14:电池的循环耐久性)对实施例1-4、实施例1-5、实施例1-8和比较例1-2的各非水电解质二次电池,在室温、3.0V~4.1V(vs.Li基准)的范围反复进行CC充放电,测定初次充放电时的放电容量、100次循环时的放电容量和500次循环时的放电容量。而且,将初次充放电时的各非水电解质二次电池的容量设为100%,计算100次循环时和500次循环时的各非水电解质二次电池的容量维持率(%)。将结果示于表18。表18如表18所示,实施例1-4、实施例1-5和实施例1-8的非水电解质二次电池尽管不含作为SEI的材料的EC,但显示了与含有EC的比较例1-2的非水电解质二次电池同等的容量维持率。认为这是由于各实施例的非水电解质二次电池中的正极和负极中存在来自本发明的电解液的含S、O被膜。而且,实施例1-4的非水电解质二次电池特别是在经过500次循环时也显示了极高的容量维持率,耐久性特别优异。根据该结果,可以说选择DMC作为有机溶剂时,与选择AN的情况相比,耐久性进一步提高。(评价例15:高温储藏试验)对实施例1-4、实施例1-5和比较例1-2的非水电解质二次电池进行在60℃储藏1周的高温储藏试验。在高温储藏试验开始前,从3.0VCC-CV(恒定电流恒定电压)充电至4.1V。以此时的充电容量为基准(SOC100),相对于该基准CC放电20%部份而调整成SOC80后,开始高温储藏试验。在高温储藏试验后以1C进行CC-CV放电至3.0V。由此时的放电容量和储藏前的SOC80容量之比,如下式那样计算残余容量。将结果示于表19。残余容量=100×(储藏后的CC-CV放电容量)/(储藏前的SOC80容量)表19实施例1-4和实施例1-5的非水电解质二次电池的残余容量比比较例1-2的非水电解质二次电池的残余容量大。根据该结果,可以说来自本发明的电解液的形成于正极和负极的含S、O被膜也有助于残余容量增大。(评价例16:倍率容量特性)用以下的方法评价实施例1-1和比较例1-1的非水电解质二次电池的倍率容量特性。将各电池的容量调整成160mAh/g。评价条件是对各非水电解质二次电池,在以0.1C、0.2C、0.5C、1C、2C的速度进行充电后进行放电,测定各速度下的工作电极的容量(放电容量)。将0.1C放电后和1C放电后的放电容量示于表20。应予说明,表20中示出的放电容量是相对于正极活性物质的质量(g)的容量。表20实施例1-1比较例1-10.1C容量(mAh/g)158.3158.21.0C容量(mAh/g)137.5125.0如表20所示,放电速度慢时(0.1C),在实施例1-1的非水电解质二次电池与比较例1-1的非水电解质二次电池之间几乎没有放电容量的差异。但是,放电速度快时(1.0C),实施例1-1的非水电解质二次电池的放电容量比比较例1-1的非水电解质二次电池的放电容量大。该结果证明了本发明的非水电解质二次电池的倍率容量特性优异。如上所述,认为这是由于本发明的非水电解质二次电池中的电解液与现有的电解液不同,另外,在本发明的非水电解质二次电池的负极和/或正极形成的含S、O被膜也与现有的不同。(评价例17:0℃、SOC20%时的输出特性评价)对上述的实施例1-1和比较例1-1的非水电解质二次电池的输出特性进行评价。评价条件是充电状态(SOC)20%、0℃、使用电压范围3V-4.2V、容量13.5mAh。SOC20%、0℃例如是像在冷藏室等使用的情况那样输出特性不易体现的区域。实施例1-1和比较例1-1的非水电解质二次电池的输出特性的评价分别是进行2秒输出和5秒输出各3次。将输出特性的评价结果示于表21。表21中的“2秒输出”是指在放电开始2秒后的输出,“5秒输出”是指在放电开始5秒后的输出。在后述的表22~表23中也是同样的。表21输出特性(0℃、SOC20%)如表21所示,实施例1-1的非水电解质二次电池在0℃、SOC20%时的输出与比较例1-1的非水电解质二次电池的输出相比,高1.2倍~1.3倍。(评价例18:25℃、SOC20%时的输出特性评价)在充电状态(SOC)20%、25℃、使用电压范围3V―4.2V、容量13.5mAh的条件下评价实施例1-1和比较例1-1的锂离子电池的输出特性。实施例1-1和比较例1-1的非水电解质二次电池的输出特性的评价分别是进行2秒输出和5秒输出各3次。将评价结果示于表22。表22输出特性(25℃、SOC20%)如表22所示,实施例1-1的非水电解质二次电池在25℃、SOC20%时的输出与比较例1-1的非水电解质二次电池的输出相比,高1.2倍~1.3倍。(评价例19:温度对输出特性的影响)另外,分析测定时的温度对上述的实施例1-1和比较例1-1的非水电解质二次电池的输出特性的影响。在0℃和25℃下测定,在任一温度下的测定中,评价条件均是充电状态(SOC)20%、使用电压范围3V―4.2V、容量13.5mAh。求出0℃下的输出相对于25℃下的输出的比率(0℃输出/25℃输出)。将该结果示于表23。表230℃输出/25℃输出实施例1-1比较例1-12秒输出0.260.275秒输出0.260.26如表23所示,对于实施例1-1的非水电解质二次电池,2秒输出和5秒输出中的0℃下的输出相对于25℃下的输出的比率(0℃输出/25℃输出)为与比较例1-1的非水电解质二次电池同等程度,可知实施例1-1的非水电解质二次电池能够与比较例1-1的非水电解质二次电池同等程度地抑制低温下的输出降低。(评价例20:热稳定性)用以下的方法评价实施例1-1、比较例1-1的非水电解质二次电池的电解液相对于充电状态的正极的热稳定性。在充电终止电压4.2V、恒定电流恒定电压条件下将非水电解质二次电池充满电。将充满电后的非水电解质二次电池解体,取出正极。将从该正极得到的正极活性物质层3mg和电解液1.8μL放入不锈钢制的锅内,密闭该锅。使用密闭锅,在氮环境下,以升温速度20℃/min的条件进行差示扫描量热分析,观察DSC曲线。作为差示扫描量热测定装置,使用RigakuDSC8230。将实施例1-1的非水电解质二次电池的充电状态的正极活性物质层与电解液共存时的DSC图表示于图61。另外,将比较例1-1的非水电解质二次电池的充电状态的正极活性物质层与电解液共存时的DSC图表分别示于图62。由图61和图62的结果可知,实施例1-1的非水电解质二次电池中的充电状态的正极与电解液共存时的DSC曲线中几乎没有观察到吸热和放热峰,与此相对,在比较例1-1的非水电解质二次电池的充电状态的正极与电解液共存时的DSC曲线中在300℃附近观察到放热峰。推断该放热峰是由正极活性物质与电解液反应的结果产生的。由这些结果可知使用了本发明的电解液的非水电解质二次电池与使用了现有的电解液的非水电解质二次电池相比较,正极活性物质与电解液的反应性低,热稳定性优异。然而,如上所述,认为酰亚胺盐容易腐蚀铝集电体。以往,使用铝集电体时,出于在该铝集电体形成用于抑制腐蚀的保护被膜的目的,认为电解液的金属盐的一部分需要使用LiPF6等锂盐。例如在日本特开2013-145732的实施例中,在电解液中配合了为酰亚胺盐的4倍左右的LiPF6。与此相对,如下所示,本发明的电解液不易腐蚀铝。因此,本发明的非水电解质二次电池中可优选使用铝集电体。(评价例21:Al的溶出确认I)(EB4)如下制造使用了电解液E8的非水电解质二次电池。将直径13.82mm、面积1.5cm2、厚度20μm的铝箔(JISA1000号)作为工作电极,对电极为金属Li。隔离件使用厚度400μm的Whatman玻璃纤维滤纸:型号1825-055。将工作电极、对电极、隔离件和E8的电解液收容在电池盒(宝泉株式会社制CR2032型纽扣电池盒)中得到非水电解质二次电池。对EB4,以1mV/s的速度在3.1V~4.6V(vs.Li基准)的范围反复进行10次线性扫描伏安法测定(所谓LSV),观察此时的电流和电极电位的变化。将表示EB4的第1次、第2次、第3次充放电的电流与电极电位的关系的图示于图63。由图63可知工作电极为Al的EB4中,在4.0V几乎没有发现电流,在4.3V电流短暂增大一点,但其后直至4.6V没有看到大幅度的增大。另外,随着充放电的反复电流量减少而趋于稳定状态。根据以上的结果,认为使用本发明的电解液且正极使用铝集电体的非水电解质二次电池即便在高电位下也不易发生Al的溶出。不易发生Al的溶出的理由尚不明确,但推测可能是本发明的电解液与现有的电解液相比金属盐和有机溶剂的种类、存在环境和金属盐浓度不同,与现有的电解液相比,Al对本发明的电解液的溶解性低。(评价例22:用工作电极Al的循环伏安法评价)(EB5)使用电解液E11代替电解液E8,除此之外,与EB4同样地得到非水电解质二次电池EB5。(EB6)使用电解液E16代替电解液E8,除此之外,与EB4同样地得到非水电解质二次电池EB6。(EB7)使用电解液E19代替电解液E8,除此之外,与EB4同样地得到非水电解质二次电池EB7。(EB8)使用电解液E13代替电解液E8,除此之外,与EB4同样地得到非水电解质二次电池EB8。(CB4)使用电解液C5代替电解液E8,除此之外,与EB4同样地得到非水电解质二次电池CB4。(CB5)使用电解液C6代替电解液E8,除此之外,与EB4同样地得到非水电解质二次电池CB5。对非水电解质二次电池EB4~EB7和CB4在3.1V~4.6V、1mV/s的条件下进行5次循环的循环伏安法评价,其后,在3.1V~5.1V、1mV/s的条件下进行5次循环的循环伏安法评价。另外,对半电池EB5、EB8和CB5在3.0V~4.5V、1mV/s的条件下进行10次循环的循环伏安法评价,其后,在3.0V~5.0V、1mV/s的条件下进行10次循环的循环伏安法评价。将表示针对EB4~EB7和CB4的电位与响应电流的关系的曲线示于图64~图72。另外,将表示针对EB5、EB8和CB5的电位与响应电流的关系的曲线示于图73~图78。由图72可知在CB4中,在2次循环以后,从3.1V到4.6V也有电流流动,随着变成高电位电流增大。另外,由图77和图78可知在CB5中也是同样的,在2次循环以后,从3.0V到4.5V也有电流流动,随着变成高电位电流增大。推断该电流是由工作电极的铝被腐蚀而产生的Al的氧化电流。另一方面,由图64~图71可知在EB4~EB7中,在2次循环以后,从3.1V到4.6V几乎没有电流流动。在4.3V以上随着电位上升观察到电流的少许增大,但随着反复循环,电流的量减少,趋于稳定状态。特别是EB5~EB7中,在为高电位的5.1V以前没有观察到电流的明显增大,并且,随着循环的反复观察到电流量的减少。另外,由图73~图76可知在EB5和EB8中也是同样的,在2次循环以后,从3.0V到4.5V几乎没有电流流动。特别是在第3次循环以后在达到4.5V以前电流几乎没有增大。而且,在EB8中,虽在为高电位的4.5V以后看到电流的增大,但其与CB5中的4.5V以后的电流值相比是非常小的值。EB5中,在4.5V以后直至达到5.0V几乎没有电流的增大,与EB5~EB7同样地,随着循环的反复观察到电流量的减少。根据循环伏安法评价的结果,可以说即便在超过5V的高电位条件下,电解液E8、E11、E16和E19对铝的腐蚀性也低。即,可以说电解液E8、E11、E16和E19对于集电体等使用了铝的电池而言是优选的电解液。(评价例23:Al的溶出确认II)使实施例1-1、实施例1-2和比较例1-1的非水电解质二次电池为使用电压范围3V~4.2V,以倍率1C反复进行100次充放电,在充放电100次后解体,取出负极。用ICP(电感耦合等离子体)发射光谱分析装置测定从正极溶出到电解液而沉淀在负极的表面的Al量。将测定结果示于表24。表24的Al量(%)是用%表示每1g负极活性物质层的Al的质量,Al量(μg/片)表示每1片负极活性物质层的Al的质量(μg),利用Al量(%)÷100×各负极活性物质层1片的质量=Al量(μg/片)的计算式进行计算。表24Al量(%)Al量(μg/片)实施例1-10.0048011.183实施例1-20.0058513.634比较例1-10.0327676.331实施例1-1和实施例1-2的非水电解质二次电池与比较例1-1的非水电解质二次电池相比,沉淀在负极表面的Al量少很多。由此可知,使用了本发明的电解液的实施例1-1和实施例1-2的非水电解质二次电池与使用了现有的电解液的比较例1-1的非水电解质二次电池相比抑制了Al从正极的集电体的溶出。(评价例24:Al集电体的表面分析)使实施例1-1和实施例1-2的非水电解质二次电池为使用电压范围3V~4.2V,以倍率1C反复进行100次充放电,在充放电100次后解体,分别取出作为正极用集电体的铝箔,用碳酸二甲酯清洗铝箔的表面。边通过Ar溅射对清洗后的实施例1-1和实施例1-2的非水电解质二次电池的铝箔的表面进行蚀刻边用X射线光电子能谱法(XPS)进行表面分析。将实施例1-1和实施例1-2的非水电解质二次电池的充放电后的铝箔的表面分析结果示于图79和图80。对比图79和图80,可知实施例1-1和实施例1-2的非水电解质二次电池的充放电后的作为正极用集电体的铝箔的表面分析结果是两者几乎相同的,阐述以下的内容。在铝箔的表面,最表面的Al的化学状态为AlF3。如果在深度方向蚀刻铝箔,则检测到Al、O、F的峰。在从表面对铝箔进行1次~3次蚀刻的位置,Al的化学状态为Al-F键和Al-O键的复合状态。如果进一步蚀刻则从4次蚀刻(以SiO2换算深度约为25nm)的位置开始O、F的峰消失,仅观察到Al的峰。应予说明,在XPS测定数据中,在Al峰位置76.3eV观察到AlF3,在Al峰位置73eV观察到纯Al,而对于Al-F键和Al-O键的复合状态,在Al峰位置74eV~76.3eV观察到。图79和图80所示的虚线表示AlF3、Al、Al2O3各自的代表性峰位置。根据以上的结果,能够确认在本发明的充放电后的非水电解质二次电池的铝箔的表面,在深度方向约25nm的厚度,形成了Al-F键(推测为AlF3)的层以及Al-F键(推测为AlF3)和Al-O键(推测为Al2O3)混合的层。也就是说,可知在正极集电体使用了铝箔的本发明的非水电解质二次电池中,即便使用本发明的电解液在充放电后也能在铝箔的最表面形成由Al-F键(推测为AlF3)构成的钝化膜。由评价例21~评价例24的结果可知在组合本发明的电解液和由铝或铝合金构成的正极用集电体的非水电解质二次电池中,通过充放电在正极用集电体的表面形成钝化膜,并且,即便在高电位状态下也抑制Al从正极用集电体的溶出。(评价例25:正极含S、O被膜分析)利用TOF-SIMS(Time-of-FlightSecondaryIonMassSpectrometry:飞行时间型二次离子质谱法),分析实施例1-4的正极含S、O被膜所含的各分子的结构信息。将实施例1-4的非水电解质二次电池在25℃进行3次循环充放电后,在3V放电状态下解体取出正极。另外地,将实施例1-4的非水电解质二次电池在25℃进行500次循环充放电后,在3V放电状态下解体取出正极。进而另外地,将实施例1-4的非水电解质二次电池在25℃进行3次循环充放电后,在60℃放置一个月,在3V放电状态下解体取出正极。用DMC将各正极清洗3次,得到分析用的正极。应予说明,在该正极形成正极含S、O被膜,通过以下的分析,对正极含S、O被膜所含的分子的结构信息进行分析。利用TOF-SIMS对分析用的各正极进行分析。作为质谱仪,使用飞行时间型二次离子质谱仪,测定正二次离子和负二次离子。使用Bi作为一次离子源,一次加速电压为25kV。作为溅射离子源,使用Ar-GCIB(Ar1500)。将测定结果示于表25~表27。应予说明,表26中的各碎片的正离子强度(相对值)是将检测到的全部碎片的正离子强度的总和设为100%的相对值。同样地,表27记载的各碎片的负离子强度(相对值)是将检测到的全部碎片的负离子强度的总和设为100%的相对值。表25(检测到的主要碎片)表26(正离子分析结果)表27(负离子分析结果)如表25所示,推断为来自电解液的溶剂的碎片仅是作为正二次离子被检测到的C3H3和C4H3。另外,推断为来自电解液的盐的碎片主要作为负二次离子被检测到,与上述的来自溶剂的碎片相比,离子强度大。此外,含有Li的碎片主要作为正二次离子被检测到,含有Li的碎片的离子强度在正二次离子和负二次离子中占很大的比例。由此推测本发明的含S、O被膜的主成分是来自电解液所含的金属盐的成分,且本发明的含S、O被膜含有大量的Li。此外,如表25所示,作为推断为来自盐的碎片,还检测到SNO2、SFO2、S2F2NO4等。它们均具有S=O结构,且是N、F与S键合的结构。也就是说,在本发明的含S、O被膜中,S不仅与O形成双键,还可以获得像SNO2、SFO2、S2F2NO4等这样与其它的元素键合的结构。因此,可以说本发明的含S、O被膜只要至少具有S=O结构即可,S=O结构所含的S可以与其它的元素键合。应予说明,当然,本发明的含S、O被膜可以含有不形成S=O结构的S和O。然而,在例如上述的日本特开2013-145732介绍的现有型的电解液,即含有作为有机溶剂的EC、作为金属盐的LiPF6和作为添加剂的LiFSA的现有电解液中,S被引入有机溶剂的分解物。因此认为S在负极被膜和/或正极被膜中以CpHqS(p、q各自为独立的整数)等离子的形式存在。与此相对,如表25~表27所示,从本发明的含S、O被膜检测到的含有S的碎片不是CpHqS碎片,反映了阴离子结构的碎片为主体。这也表明本发明的含S、O被膜与在现有的非水电解质二次电池中形成的被膜从根本上是不同的。(其它的方式I)对使用了本发明的电解液的非水电解质二次电池如下评价电池特性。(EB9)如下制造使用了电解液E8的非水电解质二次电池。将作为活性物质的平均粒径10μm的石墨90质量份和作为粘结剂的聚偏氟乙烯10质量份混合。使该混合物分散于适量的N-甲基-2-吡咯烷酮,制作浆料。准备厚度20μm的铜箔作为集电体。使用刮刀,在该铜箔的表面将上述浆料涂布成膜状。将涂布有浆料的铜箔干燥而除去N-甲基-2-吡咯烷酮,其后,对铜箔加压,得到接合物。将得到的接合物用真空干燥机在120℃加热干燥6小时,得到形成有活性物质层的铜箔。将其作为工作电极。应予说明,每1cm2铜箔的活性物质的质量为1.48mg。另外,加压前的石墨和聚偏氟乙烯的密度为0.68g/cm3,加压后的活性物质层的密度为1.025g/cm3。对电极为金属Li。将工作电极、对电极、夹在两者之间的作为隔离件的厚度400μm的Whatman玻璃纤维滤纸和电解液E8收容在直径13.82mm的电池盒(宝泉株式会社制CR2032型纽扣电池盒)中得到非水电解质二次电池EB9。(EB10)使用电解液E11,除此之外,用与EB9同样的方法,得到非水电解质二次电池EB10。(EB11)使用电解液E16,除此之外,用与EB9同样的方法,得到非水电解质二次电池EB11。(EB12)使用电解液E19,除此之外,用与EB9同样的方法,得到非水电解质二次电池EB12。(CB6)使用电解液C5,除此之外,用与EB9同样的方法,得到非水电解质二次电池CB6。(评价例26:倍率特性)用以下的方法对EB9~EB12、CB6的倍率特性进行试验。以0.1C、0.2C、0.5C、1C、2C倍率对各非水电解质二次电池进行充电后进行放电,测定各速度下的工作电极的容量(放电容量)。应予说明,1C是指在一定电流下经1小时使电池完全充电或放电所需的电流值。另外,这里的记述是将对电极看作负极,将工作电极看作正极。计算其它倍率下的容量相对于0.1C倍率下的工作电极的容量的比例(倍率特性)。将结果示于表28。表28证明了EB9、EB10、EB11、EB12在0.2C、0.5C、1C的倍率下,与CB6相比较,抑制了容量降低,进而EB9、EB10即便在2C的倍率下与CB6相比较,也抑制了容量降低,显示了优异的倍率特性。(评价例27:容量维持率)用以下的方法对EB9~EB12、CB6的容量维持率进行试验。以充放电倍率0.1C对各非水电解质二次电池进行3次如下充放电循环,即,在25℃进行CC充电(恒定电流充电)直至电压达到2.0V,再进行CC放电(恒定电流放电)直至电压达到0.01V的2.0V-0.01V的充放电循环。其后,按0.2C、0.5C、1C、2C、5C、10C的顺序针对各充放电倍率各进行3次循环的充放电,最后以0.1C进行3次循环的充放电。各非水电解质二次电池的容量维持率(%)利用以下的式子求出。容量维持率(%)=B/A×100A:最初的0.1C充放电循环中的第2次的工作电极的放电容量B:最后的0.1C的充放电循环中的第2次的工作电极的放电容量将结果示于表29。应予说明,这里的记述是将对电极看作负极,将工作电极看作正极。表29EB9EB10EB11EB12CB6容量维持率(%)98.198.798.999.898.8任一非水电解质二次电池均良好地进行了充放电反应,显示了适当的容量维持率。特别是EB10、EB11、EB12的容量维持率非常优异。(评价例28:充放电的可逆性)以充放电倍率0.1C对EB9~EB12、CB6进行3次如下充放电循环,即,在25℃进行CC充电(恒定电流充电)直至电压达到2.0V,再进行CC放电(恒定电流放电)直至电压达到0.01V的2.0V-0.01V的充放电循环。将各非水电解质二次电池的充放电曲线示于图81~图85。如图81~图85所示,可知EB9~EB12与使用了一般的电解液的CB6同样地,可逆地进行充放电反应。(EB13)使用电解液E9,除此之外,与EB9同样地得到非水电解质二次电池EB13。(评价例29:低温下的倍率特性)使用EB13和CB6,如下评价-20℃的倍率特性。将结果示于图86和图87。(1)电流向着进行锂向负极(评价电极)吸留的方向流动。(2)电压范围:2V→0.01V(v.s.Li/Li+)(3)倍率:0.02C、0.05C、0.1C、0.2C、0.5C(达到0.01V后电流停止)应予说明,1C表示在一定电流下经1小时使电池完全充电或放电所需的电流值。由图86和图87可知各电流倍率下的EB13的电压曲线与CB6的电压曲线相比较,显示出高的电压。该结果证明了本发明的非水电解质二次电池即便在低温环境下也显示优异的倍率特性。(实施例2-1)将聚丙烯酸(PAA)溶解于纯水,制备粘结剂溶液。向该粘结剂溶液添加混合鳞片状石墨粉末,制备浆料状的负极合材。浆料中的各成分(固体成分)的组成比是石墨:PAA=90:10(质量比)。使用刮刀将该浆料涂布在厚度18μm的电解铜箔(集电体)的表面,在铜箔上形成负极活性物质层。其后,在80℃干燥20分钟,使纯水从负极活性物质层蒸发除去。干燥后,利用辊压机,使集电体与负极活性物质层牢固地密合接合。将其在80℃真空干燥6小时,得到负极活性物质层的厚度为30μm左右的负极。使用上述制作的负极作为评价电极,制作非水电解质二次电池(半电池)。对电极为金属锂箔(厚度500μm)。将对电极剪裁成φ15mm,将评价电极剪裁成φ11mm,将隔离件(厚度400μm的Whatman玻璃纤维滤纸)夹在两者之间而制成电极体电池。将该电极体电池收容在电池盒(宝泉株式会社制CR2032纽扣电池)中。然后注入电解液E8,将电池盒密闭而得到实施例2-1的非水电解质二次电池。将实施例2-1的非水电解质二次电池和以下的各实施例的非水电解质二次电池的详细内容示于实施例栏的末尾的表40。(实施例2-2)使用CMC和SBR的混合物(以质量比计CMC:SBR=1:1)代替PAA作为粘结剂,并按以质量比计活性物质:粘结剂=98:2的方式使用,除此之外,与实施例2-1同样地制作负极,其余与实施例2-1同样地得到实施例2-2的非水电解质二次电池。(比较例2-1)使用与PAA同量的PVdF代替PAA作为粘结剂,除此之外,与实施例2-1同样地制作负极,其余与实施例2-1同样地得到比较例2-1的非水电解质二次电池。(比较例2-2)使用与PAA同量的PVdF代替PAA作为粘结剂,除此之外,与实施例2-1同样地制作负极。使用该负极作为评价电极,并使用电解液C5代替电解液E8,除此之外,与实施例2-1同样地得到非水电解质二次电池。使用实施例2-1和2-2以及比较例2-1和2-2的非水电解质二次电池,分别评价倍率容量特性、循环容量维持率、负荷特性。(评价例30:倍率容量)(1)电流向着进行锂向负极吸留的方向流动。(2)电压范围:2V→0.01V(v.s.Li/Li+)(3)倍率:0.1C、0.2C、0.5C、1C、2C、5C、10C、0.1C(在达到0.01V后电流停止)(4)各倍率各进行3次(合计24次循环)测定在上述的条件下测定0.1C的电流容量和各C倍率下的电流容量,求出2C倍率的电流容量相对于0.1C的电流容量的比和5C倍率的电流容量相对于0.1C的电流容量的比。将结果示于表30。应予说明,1C表示在一定电流下经1小时使电池完全充电或放电所需的电流值。(评价例31:循环容量维持率)作为循环容量维持率,计算第25次循环的电流容量相对于第1次循环的电流容量的比。将结果示于表30。表30由实施例2-1和比较例2-1的比较可知,本发明的电解液和PAA粘结剂组合与本发明的电解液和PVdF粘结剂的组合相比,循环容量维持率和高倍率侧(5C/0.1C)的负荷特性大大提高了。应予说明,由于比较例2-2的循环容量维持率高,所以认为比较例2-1中的循环容量维持率的降低现象是本发明的电解液和PVdF粘结剂的组合中特有的现象。另外由实施例2-2和比较例2-1的比较可知,本发明的电解液和CMC-SBR粘结剂的组合,与本发明的电解液和PVdF粘结剂的组合相比,也是循环容量维持率和高倍率侧(5C/0.1C)的负荷特性大大提高了。应予说明,比较例2-2中尽管使用了PVdF粘结剂但循环容量维持率高,因此认为使用本发明的电解液时,需要与粘结剂的适当组合。将实施例2-1、2-2和比较例2-1的非水电解质二次电池初次充放电曲线示于图88。根据图88,确认了在比较例2-1中在初次充电1.3V(对Li)附近产生了副反应,与此相对,在实施例2-1、2-2中由于本发明的电解液和粘结剂的适当组合抑制了副反应。由此,推测在实施例2-1、2-2中循环特性提高。抑制副反应的理由尚不确定,但可能是具有亲水基团的粘结剂产生的保护作用所引起的。另外,将实施例2-1和比较例2-1的高倍率侧(5C)的充放电曲线比较,结果在实施例2-1中发现了来自电池反应的平稳区域,与此相对,在比较例2-1中没有发现来自电池反应的平稳区域,仅靠吸附系的原理得到了微量的充电容量。由此,推测实施例2-1中负荷特性提高不仅是由于吸附容量增大,也是通过PAA粘结剂的锂供给作用使浓度过电压降低的结果。(实施例2-3)将CMC和SBR的混合物(以质量比计CMC:SBR=1:1)溶解于纯水,制备粘结剂溶液。向该粘结剂溶液添加混合石墨粉末,制备浆料状的负极合剂。浆料中的各成分(固体成分)的组成比是石墨:CMC:SBR=98:1:1(质量比)。将厚度20μm的电解铜箔作为负极用集电体,使用刮刀在该负极用集电体的表面涂布上述的浆料,在集电体上形成负极活性物质层。其后,在80℃干燥20分钟,使有机溶剂从负极活性物质层挥发除去。干燥后,利用辊压机,使负极用集电体与负极活性物质层牢固地密合接合。将其在100℃真空干燥6小时,形成负极活性物质层的单位面积重量为8.5mg/cm2左右的负极。正极活性物质层具有正极活性物质、粘结剂和导电助剂。使用NCM523作为正极活性物质,使用PVDF作为粘结剂,使用AB作为导电助剂。正极用集电体由厚度20μm的铝箔构成。将正极活性物质层设为100质量份时的正极活性物质与粘结剂与导电助剂的含有质量比为94:3:3。将NCM523、PVDF和AB以成为上述的质量比的方式混合,添加作为溶剂的NMP得到糊状的正极合剂。使用刮刀将该糊状的正极合剂涂布在正极用集电体的表面,形成正极活性物质层。通过将正极活性物质层在80℃干燥20分钟,从而通过挥发除去NMP。使用辊压机将正极活性物质层和正极用集电体的复合物压缩,使正极用集电体与正极活性物质层牢固地密合接合。将得到的接合物在120℃用真空干燥机加热6小时,切成规定的形状,得到正极。使用上述的正极、负极和电解液E8,制作作为非水电解质二次电池的一种的层压型锂离子二次电池。详细而言,在正极和负极之间夹设作为隔离件的纤维素无纺布(厚度20μm)而制成极板组。将该极板组用二片一组的层压膜覆盖,将三边进行密封后,向呈袋状的层压膜注入上述电解液。其后,将剩余的一边进行密封,由此得到四边被气密地密封、密闭极板组和电解液的实施例2-3的非水电解质二次电池。(比较例2-3)使用10质量%的PVdF代替CMC-SBR作为粘结剂,除此之外,与实施例2-3同样地制作负极,其余与实施例2-3同样地得到比较例2-3的非水电解质二次电池。(比较例2-4)使用电解液C5代替电解液E8,除此之外,与实施例2-3同样地得到比较例2-4的非水电解质二次电池。(比较例2-5)将作为负极活性物质的天然石墨90质量份和作为粘结剂的PVdF10质量份混合。使该混合物分散于适量的离子交换水,得到浆料状的负极合剂。准备厚度20μm的铜箔作为负极用集电体。使用刮刀,在该负极用集电体的表面将上述负极合剂涂布成膜状。将负极合剂和负极用集电体的复合物干燥而除去水,其后进行加压,得到接合物。将得到的接合物用真空干燥机在120℃加热干燥6小时,得到在负极用集电体上形成有负极活性物质层的负极。正极与实施例2-3的非水电解质二次电池的正极同样地进行制造。除使用了该正极、负极和电解液C5以外,与实施例2-3同样地得到比较例2-5的非水电解质二次电池。(评价例32:输入输出特性)使用实施例2-3和比较例2-3~2-5的非水电解质二次电池,按以下的条件评价输入(充电)特性。(1)使用电压范围:3V-4.2V(2)容量:13.5mAh(3)SOC80%(4)温度:0℃、25℃(5)测定次数:各3次评价条件是充电状态(SOC)80%、0℃、25℃、使用电压范围3V-4.2V、容量13.5mAh。SOC80%、0℃例如是像在冷藏室等使用的情况那样输入特性不易体现的区域。实施例2-3和比较例2-3、2-4的输入特性的评价是分别进行3次2秒输入和5秒输入。将输入特性的评价结果示于表31、表32。表中的“2秒输入”表示在充电开始2秒后的输入,“5秒输入”表示在充电开始5秒后的输入。应予说明,表31、32中,将实施例2-3和比较例2-3中使用的电解液E8简称为“FSA”,将比较例2-4和比较例2-5中使用的电解液C5简称为“ECPF”。表31(25℃SOC80%)表32(0℃SOC80%)在0℃和25℃这两个温度下,实施例2-3与比较例2-3~2-5相比,输入(充电)特性提高。这是并用了具有亲水基团的粘结剂(CMC-SBR)和本发明的电解液所产生的效果,特别是即便在0℃下也显示高的输入(充电)特性,因此表明了即便在低温下电解液中的锂离子的移动也能顺利地进行。(实施例2-4)使用CMC和SBR的混合物(以质量比计CMC:SBR=1:1)代替PAA作为粘结剂,并按以质量比计活性物质:粘结剂=98:2的方式使用,且使真空干燥温度为100℃,除此之外,与实施例2-1同样地形成负极活性物质层的单位面积重量为4mg/cm2左右的负极。使用NCM523作为正极活性物质,使用PVDF作为粘结剂,使用AB作为导电助剂。作为正极用集电体,使用厚度20μm的铝箔。将正极活性物质层设为100质量份时的正极活性物质与导电助剂与粘结剂的含有质量比为90:8:2。使用这些正极活性物质、导电助剂、粘结剂和正极用集电体,与实施例2-3同样地得到正极。使用上述的正极、负极和上述的电解液E11,与实施例2-3同样地得到实施例2-4的非水电解质二次电池。(比较例2-6)使用电解液C5代替电解液E11,除此之外,与实施例2-4同样地得到比较例2-6的非水电解质二次电池。(评价例33:电池的循环耐久性)使用实施例2-4和比较例2-6的非水电解质二次电池,各自进行500次反复循环试验,即,在温度25℃、1C的CC充电的条件下充电至4.1V,休止1分钟后,以1C的CC放电,放电至3.0V,休止1分钟的循环。将测定第500次循环的放电容量维持率的结果示于表33。放电容量维持率是由用第500次循环的放电容量除以初次的放电容而得的值的百分比{(第500次循环的放电容量)/(初次的放电容量)×100}求出的值。另外,在第200次循环中,由以温度25℃、0.5C的CCCV调整至电压3.5V后、以3C进行10秒的CC放电时的电压变化量(放电前电压与放电10秒后电压的差)和电流值利用欧姆定律测定直流电阻。将结果示于表33。表33E11:3.9MLiFSA/DMC、C5:1.0MLiPF6/EC+DEC如实施例2-4所示,通过组合由具有亲水基团的聚合物构成的粘结剂与本发明涉及的本发明电解液,能够制成循环寿命提高且低电阻的二次电池。(实施例2-5)按以质量比计活性物质:粘结剂=90:10的方式使用PAA代替CMC-SBR,除此之外,与实施例2-4同样地制作负极,使用该负极,除此之外,与实施例2-4同样地得到实施例2-5的非水电解质二次电池。(评价例34:电池的高温储藏耐性)使用实施例2-4、2-5、比较例2-6的锂二次电池,进行在60℃储藏1周的高温储藏试验。在高温储藏试验开始前,以通过CC-CV从3.0V充电至4.1V时的充电容量为基准(SOC100),相对于基准CC放电20%(调整成SOC80)后,开始高温储藏试验。在高温储藏试验后以1CCC-CV至3.0V,根据此时的放电容量与储藏前的SOC80容量之比,按下式计算保留容量。将结果示于表34。保留容量=100×(储藏后的CC-CV放电容量)/(储藏前的SOC80容量)表34E11:3.9MLiFSA/DMC、C5:1.0MLiPF6/EC+DEC如实施例2-4、2-5所示,通过组合由具有亲水基团的聚合物构成的粘结剂与本发明涉及的本发明电解液,高温储藏后的容量提高。(评价例35:电池的循环耐久性)在室温、3.0V~4.1V(vs.Li基准)的范围对实施例2-4和比较例2-6的各非水电解质二次电池反复进行500次循环CC充放电,测定各循环中的放电电流容量(Ah)和充电电流容量(Ah)。然后,基于测定值计算各循环中的库仑效率(%),进一步计算从初次充放电时(也就是说1次循环时)到500次循环时的库仑效率的平均值。另外,测定初次充放电时的放电容量和500次循环时的放电容量。然后,将初次充放电时的各非水电解质二次电池的容量设为100%,计算500次循环时的各非水电解质二次电池的容量维持率(%)。库仑效率根据{(放电电流容量)/(充电电流容量)}×100计算。将结果示于表35。表35E11:3.9MLiFSA/DMC、C5:1.0MLiPF6/(EC/DEC)如表35所示,实施例2-4的非水电解质二次电池与比较例2-6的非水电解质二次电池相比,库仑效率高,容量维持率也高。也就是说,组合作为金属盐的LiFSA和作为粘结剂的CMC-SBR的情况与组合作为金属盐的LiPF6和作为粘结剂的CMC-SBR的情况相比,能提高非水电解质二次电池的循环特性。更具体而言,使用具有亲水基团的聚合物作为粘结剂的本发明的非水电解质二次电池中,可优选使用LiFSA作为电解液的金属盐。应予说明,如果负极中的副反应(也就是说电解质的分解等电池反应以外的反应)减少则库仑效率有升高的趋势。负极中的副反应大多是在负极中不可逆地捕捉Li的不可逆反应,会成为电池容量降低的原因。因此,推测在实施例4的各非水电解质二次电池中抑制了上述的副反应,其结果,500次循环时的容量维持率提高。仅限于参考,表35所示的库仑效率为500次循环的平均值,也就是说为每次循环的值。因此,若将500次循环值累积,则实施例2-4与比较例2-6的库仑效率之差是非常大的。(实施例2-6)使用与实施例2-4相同的正极(NCM523:AB:PVdF=90:8:2)和与实施例2-1相同的负极(天然石墨:PAA=90:10),除此之外,与实施例2-3同样地得到实施例2-6的非水电解质二次电池。(实施例2-7)使用与实施例2-4相同的正极(NCM523:AB:PVdF=90:8:2)和与实施例2-2相同的负极(天然石墨:CMC:SBR=98:1:1),除此之外,与实施例2-3同样地得到实施例2-7的非水电解质二次电池。(比较例2-7)使用电解液C5,除此之外,用与实施例2-6同样的方法得到比较例2-7的非水电解质二次电池。(比较例2-8)使用电解液C5,除此之外,用与实施例2-7同样的方法得到比较例2-8的非水电解质二次电池。(评价例36:电池的循环耐久性)用与上述的“评价例33:电池的循环耐久性”同样的方法对实施例2-6、2-7的各非水电解质二次电池反复进行200次循环充放电,计算200次循环时的各非水电解质二次电池的容量维持率(%)和库仑效率(%,200次循环的平均值)。将结果示于表36。表36E8:4.5MLiFSA/AN如表36所示,实施例2-6的非水电解质二次电池与实施例2-7的非水电解质二次电池相比,容量维持率和库仑效率优异。根据该结果,可以说作为粘结剂,更优选为PAA。(评价例37:电池的循环耐久性)与上述的“评价例36:电池的循环耐久性”大致同样地,对实施例2-6、2-7和比较例2-7、2-8的各非水电解质二次电池计算203次循环时的各非水电解质二次电池的容量维持率(%)。更具体而言,在该试验中,以第3次循环为试验初始,求出从此次循环再进行200次循环充放电时的容量维持率。另外,在试验初始,也就是3次循环时,由以温度25℃、0.5C的CCCV调整成电压3.5V后、以3C进行10秒的CC放电时的电压变化量(放电前电压与放电10秒后电压的差)和电流值利用欧姆定律测定直流电阻。然后,将此时的直流电阻作为初始直流电阻。将结果示于表37。表37E8:4.5MLiFSA/AN、C5:1.0MLiPF6/(EC/DEC)如表37所示,在实施例2-6、实施例2-7、比较例2-7和比较例2-8的各非水电解质二次电池中,203次循环时的容量维持率大致相同,均为高值。对实施例2-6和2-7进行比较,可以说作为粘结剂,PAA优异,对比较例2-7和2-8进行比较,可以说作为粘结剂,CMC-SBR优异。也就是说,在使用了本发明电解液的本发明的非水电解质二次电池中,作为粘结剂,可以说与使用CMC-SBR相比,更优选使用PAA。应予说明,使用LiFSA作为金属盐的实施例2-6和实施例2-7的非水电解质二次电池与使用LiPF6作为金属盐的比较例2-6和比较例2-7的非水电解质二次电池相比,初始直流电阻低。因此,为了兼具容量维持率的提高和电阻增大的抑制,可以说使用本发明的电解液且使用具有亲水基团的粘结剂作为粘结剂的实施例2-6和实施例2-7的非水电解质二次电池、即本发明的非水电解质二次电池是有利的。(评价例38:电池的高温储藏耐性)使用实施例2-6、2-7、比较例2-7,2-8的非水电解质二次电池,进行在60℃储藏1周的高温储藏试验。在高温储藏试验开始前,以通过CC-CV从3.0V充电到4.1V时的充电容量为基准,即设为SOC100。然后,相对于基准CC放电20%调整成SOC80后,开始高温储藏试验。在高温储藏试验后,以1C进行CC-CV至3.0V,基于此时的放电容量与储藏前的SOC80容量之比,通过下式计算残余容量。残余容量=100×(储藏后的CC-CV放电容量)/(储藏前的SOC80容量)计算保留容量。将结果示于表38。表38E8:4.5MLiFSA/AN、C5:1.0MLiPF6/(EC/DEC)如表38所示,实施例2-6的非水电解质二次电池与实施例2-7的非水电解质二次电池相比,残余容量大。也就是说,组合LiFSA/AN和PAA的实施例2-6的非水电解质二次电池与组合LiFSA/AN和CMC-SBR的实施例2-7的非水电解质二次电池相比,高温储藏特性优异。另外,根据该结果,可知组合本发明的电解液和由具有亲水基团的聚合物构成的粘结剂的本发明的非水电解质二次电池具有与组合通常的电解液和由具有亲水基团的聚合物构成的粘结剂的现有的非水电解质二次电池同等或同等以上的高温储藏耐性。(其它的方式II)作为本发明的电解液,具体举出以下的电解液。应予说明,以下的电解液也包括已经叙述的电解液。(电解液A)如下制造本发明的电解液。将作为有机溶剂的1,2-二甲氧基乙烷约5mL放入具备搅拌子和温度计的烧瓶中。在搅拌条件下,以溶液温度保持在40℃以下的方式向上述烧瓶中的1,2-二甲氧基乙烷缓慢地添加作为锂盐的(CF3SO2)2NLi,使其溶解。由于在加入约13g的(CF3SO2)2NLi的时刻(CF3SO2)2NLi的溶解暂时停滞,所以将上述烧瓶投入恒温槽,将烧瓶内的溶液温度加温至50℃,使(CF3SO2)2NLi溶解。由于在加入约15g的(CF3SO2)2NLi的时刻(CF3SO2)2NLi的溶解再次停滞,所以用滴管滴加1滴1,2-二甲氧基乙烷,之后(CF3SO2)2NLi溶解。进一步缓慢地添加(CF3SO2)2NLi,加入全部的规定的(CF3SO2)2NLi。将得到的电解液移至20mL容量瓶,加入1,2-二甲氧基乙烷直至容积变成20mL。得到的电解液的容积为20mL,该电解液所含的(CF3SO2)2NLi为18.38g。将其作为电解液A。电解液A中的(CF3SO2)2NLi的浓度为3.2mol/L,密度为1.39g/cm3。密度是在20℃测定的。应予说明,上述制造是在非活性气体环境下的手套箱内进行的。(电解液B)用与电解液A同样的方法,制造(CF3SO2)2NLi的浓度为2.8mol/L、密度为1.36g/cm3的电解液B。(电解液C)将作为有机溶剂的乙腈约5mL放入具备搅拌子的烧瓶中。在搅拌条件下,向上述烧瓶中的乙腈缓慢地添加作为锂盐的(CF3SO2)2NLi,使其溶解。加入规定的(CF3SO2)2NLi后,搅拌一晚。将得到的电解液移至20mL容量瓶,加入乙腈直至容积变成20mL。将其作为电解液C。应予说明,上述制造是在非活性气体环境下的手套箱内进行的。电解液C中(CF3SO2)2NLi的浓度为4.2mol/L,密度为1.52g/cm3。(电解液D)用与电解液C同样的方法,制造(CF3SO2)2NLi的浓度为3.0mol/L、密度为1.31g/cm3的电解液D。(电解液E)使用环丁砜作为有机溶剂,除此之外,用与电解液C同样的方法,制造(CF3SO2)2NLi的浓度为3.0mol/L、密度为1.57g/cm3的电解液E。(电解液F)使用二甲基亚砜作为有机溶剂,除此之外,用与电解液C同样的方法,制造(CF3SO2)2NLi的浓度为3.2mol/L、密度为1.49g/cm3的电解液F。(电解液G)使用(FSO2)2NLi作为锂盐,使用1,2-二甲氧基乙烷作为有机溶剂,除此之外,用与电解液C同样的方法,制造(FSO2)2NLi的浓度为4.0mol/L、密度为1.33g/cm3的电解液G。(电解液H)用与电解液G同样的方法,制造(FSO2)2NLi的浓度为3.6mol/L、密度为1.29g/cm3的电解液H。(电解液I)用与电解液G同样的方法,制造(FSO2)2NLi的浓度为2.4mol/L、密度为1.18g/cm3的电解液I。(电解液J)使用乙腈作为有机溶剂,除此之外,用与电解液G同样的方法,制造(FSO2)2NLi的浓度为5.0mol/L、密度为1.40g/cm3的电解液J。(电解液K)用与电解液J同样的方法,制造(FSO2)2NLi的浓度为4.5mol/L、密度为1.34g/cm3的电解液K。(电解液L)将作为有机溶剂的碳酸二甲酯约5mL放入具备搅拌子的烧瓶中。在搅拌条件下,向上述烧瓶中的碳酸二甲酯缓慢地添加作为锂盐的(FSO2)2NLi,使其溶解。加入总量为14.64g的(FSO2)2NLi后,搅拌一晚。将得到的电解液移至20mL容量瓶,加入碳酸二甲酯直至容积变成20mL。将其作为电解液L。应予说明,上述制造是在非活性气体环境下的手套箱内进行的。电解液L中的(FSO2)2NLi的浓度为3.9mol/L,电解液L的密度为1.44g/cm3。(电解液M)用与电解液L同样的方法,制造(FSO2)2NLi的浓度为2.9mol/L、密度为1.36g/cm3的电解液M。(电解液N)将作为有机溶剂的碳酸甲乙酯约5mL放入具备搅拌子的烧瓶中。在搅拌条件下,向上述烧瓶中的碳酸甲乙酯缓慢地添加作为锂盐的(FSO2)2NLi,使其溶解。加入总量为12.81g的(FSO2)2NLi后,搅拌一晚。将得到的电解液移至20mL容量瓶,加入碳酸甲乙酯直至容积变成20mL。将其作为电解液N。应予说明,上述制造是在非活性气体环境下的手套箱内进行的。电解液N中的(FSO2)2NLi的浓度为3.4mol/L,电解液N的密度为1.35g/cm3。(电解液O)将作为有机溶剂的碳酸二乙酯约5mL放入具备搅拌子的烧瓶中。在搅拌条件下,向上述烧瓶中的碳酸二乙酯缓慢地添加作为锂盐的(FSO2)2NLi,使其溶解。加入总量为11.37g的(FSO2)2NLi后,搅拌一晚。将得到的电解液移至20mL容量瓶,加入碳酸二乙酯直至容积变成20mL。将其作为电解液O。应予说明,上述制造是在非活性气体环境下的手套箱内进行的。电解液O中的(FSO2)2NLi的浓度为3.0mol/L,电解液O的密度为1.29g/cm3。表39中示出上述电解液的一览表。表39LiTFSA:(CF3SO2)2NLi、LiFSA:(FSO2)2NLi、AN:乙腈、DME:1,2-二甲氧基乙烷、DMSO:二甲基亚砜、SL:环丁砜、DMC:碳酸二甲酯、EMC:碳酸甲乙酯、DEC:碳酸二乙酯表40当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1