化合物半导体衬底的制作方法
本申请是申请日为2012年5月9日、国际申请号为pct/jp2012/061815、中国申请号为201280014248.2的专利申请的分案申请。本发明涉及清洁化合物半导体衬底的方法和化合物半导体衬底,特别地涉及清洁由例如gaas、inp或gap构成的iii-v族化合物半导体衬底的方法。
背景技术:
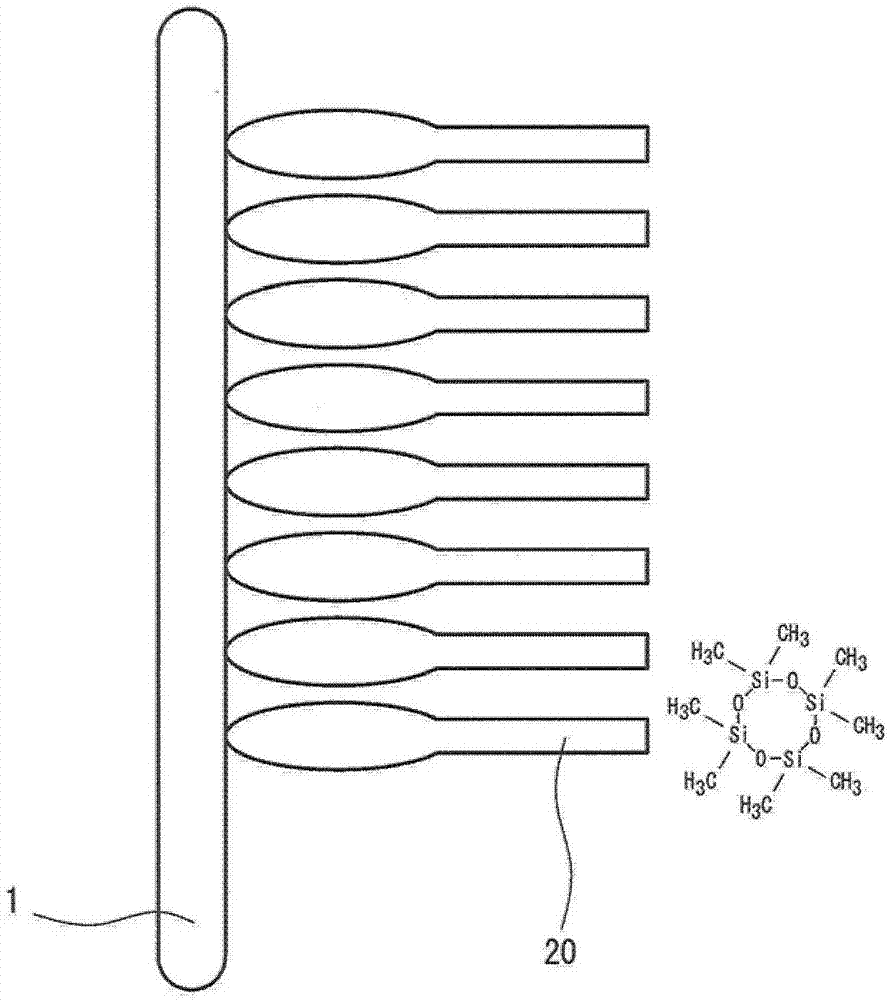
:通常,对iii-v族化合物半导体晶体、特别是例如gaas、inp或gap的化合物半导体单晶的衬底进行用氯类抛光剂镜面抛光该衬底的至少一个主表面的加工,随后用酸或碱清洁且最后用超纯水冲洗并干燥。例如,专利文献1描述了如下清洁方法,其中在被抛光的gaas衬底的表面形成天然氧化层且利用具有1ppm以下的溶解氧含量的超纯水将天然氧化层从gaas衬底的表面溶解除去。专利文献2陈述了使衬底的表面氧化且然后将该衬底浸渍在氨水、氢氧化钠的水溶液、磷酸、盐酸或氢氟酸中。专利文献3描述了如下清洁方法,其中将衬底表面上的有机物质和金属除去且然后利用酸性溶液对衬底的氧化膜进行蚀刻;然后将衬底用碱性水溶液清洁,随后用超纯水清洁且然后干燥。根据该方法,可以完全除去外来物质如析出物。现有技术文献[专利文献][专利文献1]日本特开平10-079363号公报[专利文献2]日本特开平07-211688号公报[专利文献3]日本特开2000-340535号公报技术实现要素:[技术问题]虽然gaas衬底具有高电子迁移率的优点,但在该衬底上的氧化膜不是像si衬底上的sio2膜那样的电绝缘膜。因此,当利用gaas衬底制造电子器件时,需要在除元件操作区之外的区域具有高电阻而使得电流不流过。例如,图1示出用作移动电话用电子器件的高电子迁移率晶体管(hemt)的示意性截面图。在该hemt中,在具有高电阻的gaas衬底1上形成未掺杂的高纯度外延层2,且在未掺杂的高纯度外延层2上形成电子流过的电子供给层3。电流(电子)仅旨在流动(11)通过未掺杂层2与电子供给层3之间的界面。然而,电子供给层下面的未掺杂层2具有约1μm的非常小的厚度。因此,当未掺杂层2与衬底1之间的界面或衬底1具有使得电通过的趋势时,存在认为会流动的源-漏电流越过未掺杂层2流动(12)通过未掺杂层2与衬底1之间的界面或通过衬底的情况,且结果,造成器件的电特性异常。当通过有机金属化学气相淀积(mocvd)等在已经通过现有清洁方法清洁的化合物半导体衬底的表面上生长外延膜而形成hemt时,存在附着到衬底表面的杂质如si残留在衬底与未掺杂层之间的界面处的情况,且结果,造成电特性异常。特别地,并入到iii-v族化合物半导体晶体如gaas晶体中的si充当n-型杂质以显著减小晶体的电阻。为了防止该现象,通常,在外延反应器中将外延生长之前的衬底在高温下保持一定的时间,从而使附着到衬底表面的杂质分解或蒸发(热清洁)。然而,这种方法可能造成如下问题:外延生长时间增加而降低设备生产率;和当衬底过度地暴露于高温时,衬底本身分解或蒸发而使得形成表面不规则且不能形成期望的外延层。还存在如下方法,其中,为了使界面处的si为惰性的,使碳(c)或氧(o)附着到表面。然而,该方法具有如下问题:其使用范围(条件)有限,因为当不控制si的附着量时,在外延生长之后界面的电特性变得不稳定;且当杂质蒸发的平衡由于在外延生长反应器中在热清洁时的气氛差异而变化时,可能造成电特性异常。本发明人研究了外延层与衬底之间的界面处的电阻减小的原因,且结果发现,当将已经清洁的衬底长时间储存时,造成许多上述电特性异常。为了评价,在将具有1×1015~2×1016/cm3的晶体中的碳浓度且具有1×107ωcm~6×108ωcm的电阻率的半绝缘的gaas镜面衬底清洁之后即刻,本发明人形成1-μm未掺杂的gaas外延膜而在外延生长之前不进行热清洁,从而容易地认定杂质对衬底表面的影响。利用在表面上使用的涡流探头对具有外延膜的该衬底测定外延膜的表面的方块电阻。结果,当使用清洁之后即刻的衬底时,方块电阻为4~80×104ω/□。相比之下,当使用置于聚丙烯(pp)容器中、密封在氮气中、然后密封在铝涂布的塑料袋中且储存20天以上的衬底时,方块电阻减小到3~10×103ω/□。然后,利用磁场二次离子质谱仪(sims),通过自外延膜的表面侧进行溅射对外延膜与衬底之间的界面进行元素分析。结果发现,在清洁之后即刻的衬底与已经储存20天以上的衬底之间在si含量方面存在差异。也就是说,已经储存20天以上的衬底具有比清洁之后即刻的衬底高一个或多个数量级的si含量。具体地,在清洁之后即刻进行外延生长的情况下界面处的si浓度为约1×1017原子/cm3,而发现在储存20天之后进行外延生长的情况下界面处的si浓度为约10×1017原子/cm3。这可能是电阻值异常的原因。应注意,所述si浓度指示在sims分析中在外延膜与衬底之间的界面处测定的si浓度的最大值(峰值浓度),其中重复通过溅射在深度方向上进行特定距离的蚀刻且对各种元素进行浓度分析的处理,从而分析在从外延膜的表面至衬底的深度方向上的si浓度。si浓度的最大值(峰值浓度)可能变得低于实际值,因为峰形状由于外延膜表面的粗糙化或由溅射引起的粗糙化而变宽。因此,表v描述了在界面处或在界面附近检测的si的峰值浓度以及积分浓度(薄层浓度)。在界面处或在界面附近检测的si的积分浓度指示外延晶片的单位面积(cm2)的在界面处或在界面附近存在的si原子的数目。因此,峰值浓度的误差可以用积分浓度补偿。基于上述发现,本发明的目的在于提供化合物半导体衬底和所述化合物半导体衬底的表面处理方法(在下文中,可以称为“清洁方法”),其中即使在将处理过的衬底长时间储存之后也不会出现电阻值异常。即使在将根据本发明的化合物半导体衬底长时间储存且然后在其上形成外延膜时,也不会出现电特性异常。[解决问题的手段]本发明人对于如何实现该目的进行了透彻的研究。结果,本发明人认为,在清洁室内的空气中si以约0.5μg/cm3的量作为有机物质存在,这作为分子污染不可忽略;考虑到在暴露于其中悬浮有含si有机物质(硅氧烷)的气氛的衬底和储存容器上的含si有机物质的分子数目以及在包装以发货时通过氮气吹扫未吹扫且残留在衬底上的含si有机物质的分子数目,难以完全防止含si有机物质附着到化合物半导体衬底的表面上。这实际上得到了调查且难以通过改善储存气氛和包装方法而克服由si附着引起的方块电阻的下降。如图2b中所示,硅氧烷及其他有机污染物21附着到衬底1的表面上。通常,悬浮在清洁室内的气氛中的含si有机物质主要是在三聚物(d3)~七聚物(d7)范围且具有约200~约800的分子量的分子。用酸或碱处理之后即刻的化合物半导体衬底具有活化的表面且悬浮在气氛中的物质非常可能附着到该表面上。特别地,关于要残留在界面处的si,含si有机物质可能具有适合附着到表面的结构。因为含si有机物质具有约200~约800的相对高的分子量,所以可能倾向于通过置换具有小质量数且在清洁之后即刻附着到表面的分子如丙酮和乙醇而附着到表面。另外,因为含si有机物质具有相对高的耐热性,所以在外延生长前即刻在外延反应器内进行的热清洁时,在外延生长之前在衬底表面上存在的含si有机物质不太可能分解。由于这个原因,含si有机物质可能倾向于残留在外延层与衬底之间的界面处。因此,本发明人认为,可以通过在悬浮在气氛中的含si有机物质到达表面之前形成由比含si有机物质重、易于附着到化合物半导体衬底的表面上且可以在外延膜形成时通过加热容易地蒸发的物质构成的表面层而在所制造的hemt中消除电特性异常的出现。本发明因此充分地制作了许多试样,且结果发现了本发明。根据本发明的清洁化合物半导体衬底的方法包括:用例如酸或碱清洁所述衬底,然后用超纯水冲洗所述衬底,干燥所述衬底和随后将所述衬底浸渍在非离子表面活性剂中且然后干燥所述衬底的最终步骤。已经发现,在处理已经用酸或碱清洁的化合物半导体衬底的表面的情况下,使用阴离子表面活性剂提供展示亲水性的衬底表面,而使用非离子表面活性剂提供展示疏水性的衬底表面。与阴离子表面活性剂和阳离子表面活性剂不同,非离子表面活性剂的亲水性基团不是由各自带正电或带负电的表面活性剂分子构成,而是所述亲水性基团通过与通常被称为表面活性剂分子中的亲水性基团的部分形成水分子的氢键而形成,所述部分具有碳-氧-碳键(-c-o-c-)。总之,这种非离子表面活性剂在长分子末端具有许多碳-氧键。因为附着了非离子表面活性剂的衬底展示疏水性,所以所述衬底表面和非离子表面活性剂的亲水性部分可能经由氢键结合,且由于存在许多用于氢键的连接键,所以预期比通常的附着更强的结合。非离子表面活性剂优选具有700以上且2000以下的分子量。这种表面活性剂相对易于得到且因此在工业上有利。优选在弱酸性条件:2以上且7以下的ph下进行处理。最后,将衬底浸渍在所述表面活性剂中且然后干燥,使得如图2a中所示,在化合物半导体衬底1的表面上形成一层表面活性剂层20。上述描述描述了采用非离子表面活性剂的实例。然而,所述处理不必限于非离子表面活性剂的使用,且使用具有700以上分子量的有机物质可以提供类似的优点。本发明提供一种化合物半导体衬底,其至少一个主表面被镜面抛光,所述镜面抛光的表面被含有氢(h)、碳(c)和氧(o)的有机物质覆盖。在通过根据本发明的表面处理方法提供的衬底中,覆盖表面且含有氢(h)、碳(c)和氧(o)的有机物质(表面活性剂)的层优选具有1.5nm以上且3.0nm以下的厚度。表面活性剂主要由单键构成且因此非常可能分解。因此,表面活性剂易于通过加热到100℃以上而分解并蒸发。在通过mocvd等形成外延膜时,通常在初始阶段进行600℃~700℃下的加热且因此表面活性剂在此阶段蒸发;然而,为了抑制加热时的表面粗糙化,所述加热优选以使得温度不超过生长温度550℃的方式进行,这是优选的,因为在本发明中覆盖表面的有机物质层在生长温度550℃下容易蒸发。在通过根据本发明的处理方法提供的衬底的表面中,在采用69ga+作为一次离子的飞行时间二次离子质谱(tof-sims)分析中具有31的质量数的阳离子(ch3o+)的相对信号强度优选为2.4×10-3以上。在采用69ga+作为一次离子的tof-sims分析中具有73的质量数的阳离子(c3h5o2+)的相对信号强度优选为3.2×10-4以上。在采用69ga+作为一次离子的tof-sims分析中具有31的质量数的阳离子(ch3o+)的相对强度优选为标准清洁的化合物半导体衬底的该相对强度的2.0倍以上。在采用69ga+作为一次离子的tof-sims分析中具有73的质量数的阳离子(c3h5o2+)的相对强度优选为标准清洁的化合物半导体衬底的该相对强度的4.1倍以上。在通过根据本发明的处理方法提供的衬底的表面中,在通过入射x射线能量为365ev且光电子飞离角为90°(检测在垂直于晶片表面的方向上飞离的光电子)的同步辐射x射线光电子能谱(xps)分析中,在比c1s:285ev的峰高1.5±0.5ev的高能量侧检测到峰。所述高能量侧的峰的积分强度优选为所述285ev附近的峰的积分强度的0.25倍以上。通过根据本发明的处理方法提供的衬底为其至少一个主表面被镜面抛光的化合物半导体衬底,其中,在550℃的生长温度下生长而在所述生长之前未进行热清洁的外延膜与所述化合物半导体衬底之间的界面处的硅(si)含量,以通过sims深度方向分析测定的峰值浓度计,为2×1017原子/cm3以下。[发明有利效果]根据本发明,即使将化合物半导体衬底长时间储存且然后在其上形成外延膜,也不会出现电特性异常。[附图说明][图1]图1为hemt的说明图。[图2a]图2a示出在化合物半导体衬底的表面上形成表面活性剂层。[图2b]图2b示出表面污染模型。[图3]图3示出现有表面处理方法和根据本发明的方法。[图4]图4示出非离子表面活性剂的实例。[图5]图5示出非离子表面活性剂的其他实例。[图6]图6示出在具有31的质量数的阳离子(ch3o+)的相对强度与界面si峰值浓度之间的关系。[图7]图7示出在具有73的质量数的阳离子(c3h5o2+)的相对强度与界面si峰值浓度之间的关系。[图8]图8示出同步辐射xps光谱的实例。[图9]图9示出在由同步辐射xps测定计算的积分强度与界面si浓度之间的关系。[具体实施方式]根据本发明的清洁化合物半导体衬底的方法包括:在最终步骤中,将所述衬底浸渍在包含含有氢(h)、碳(c)和氧(o)的有机物质(非离子表面活性剂)的溶液中且然后干燥所述衬底。在所述最终步骤之前的清洁方法可以以与现有方法相同的方式进行。典型地,如图3中所示,进行如下步骤:晶片清洁溶液(有机碱性溶液)→超纯水冲洗→晶片清洁溶液→超纯水冲洗→酸清洁→超纯水冲洗→超纯水冲洗→干燥;和最后,表面活性剂→超纯水冲洗,然后干燥。或者,可以消除中间干燥且可以进行如下步骤:晶片清洁溶液(有机碱性溶液)→超纯水冲洗→晶片清洁溶液→超纯水冲洗→酸清洁→超纯水冲洗→超纯水冲洗→表面活性剂→超纯水冲洗,然后干燥。通过使用具有低浓度的表面活性剂水溶液,可以消除在表面活性剂处理之后的超纯水冲洗。具体地,可以进行如下步骤:晶片清洁溶液(有机碱性溶液)→超纯水冲洗→晶片清洁溶液→超纯水冲洗→酸清洁→超纯水冲洗→超纯水冲洗→表面活性剂,然后干燥。或者,可以进行如下步骤:晶片清洁溶液(有机碱性溶液)→超纯水冲洗→晶片清洁溶液→超纯水冲洗→酸清洁→超纯水冲洗→超纯水冲洗→干燥;和最后,表面活性剂,然后干燥。所述清洁可以通过如下进行:布置包含清洁溶液、超纯水和表面活性剂的清洁槽,用所述槽依次处理化合物半导体衬底且最终通过例如使用高速旋转时的离心力进行旋转干燥而干燥所述衬底。或者,可以采用单晶片加工,其中将各衬底在水平保持的同时进行旋转,用表面活性剂和超纯水处理,且最终通过高速旋转干燥。或者,可采用分批加工,其中对衬底进行超纯水冲洗,然后从包含表面活性剂的纯水中取出并干燥。通过各种方法对由此清洁的化合物半导体衬底的表面状态进行分析:一种方法是tof-sims分析。使用由美国的物理电子公司(physicalelectronics,inc.)制造的tof-sims设备(trifii)来辐射作为一次离子的69ga+并用飞行时间质谱仪检测带正电的二次离子。质量数检测范围为0.5~2000。在tof-sims分析中,ga+一次离子的辐射引起附着分子分解且检测到分解后的分子物种。因此,具有1000以上的质量数的分子物种的检测量非常少。对相对强度进行比较,所述相对强度各自通过限定在1~1000的质量数范围内检测到的带正电的二次离子的总计数作为分母并通过限定目标质量数的计数作为分子来确定。结果,发现在附着到表面的si的含量低且方块电阻高的衬底中,具有31的质量数的阳离子(ch3o+)的相对信号强度为2.4×10-3以上;还发现具有73的质量数的阳离子(c3h5o2+)的相对信号强度为3.2×10-4以上。对已经通过现有清洁方法(标准清洁)清洁的化合物半导体衬底类似地进行tof-sims分析。将这些结果与本发明进行比较。在本发明中31的质量数的相对信号强度为标准清洁产品的相对信号强度的2.0倍以上。在本发明中73的质量数的相对信号强度为标准清洁产品的相对信号强度的4.1倍以上。“标准清洁产品”可以例如为通过图3中所示的现有方法处理的衬底。或者,其可以为通过后述的实施例1中进行的标准清洁处理的衬底。进行另一分析方法,其为采用同步辐射作为入射光的xps分析。当在入射x射线能量为365ev且光电子飞离角为90°的条件下对横轴进行校准,使得c1s的低能量侧的峰在285ev下时,在标准清洁产品中仅检测到在所述285ev下的峰,而在根据本发明的清洁产品中还检测到在比其高1~2ev的高能量侧的另一个峰。发现,当所述高能量侧的峰的积分强度为在所述285ev下的峰的积分强度的0.25倍以上时,si附着含量低且方块电阻高。代表用于表面处理且包含含有氢(h)、碳(c)和氧(o)的有机物质的溶液的非离子表面活性剂的实例包括高级醇类表面活性剂和具有700~2000的分子量的烷基酚类表面活性剂,诸如聚氧化烯烷基醚、聚氧化乙烯烷基醚和聚氧化乙烯烷基苯基醚。非离子表面活性剂可以为脂肪酸类表面活性剂。脂肪酸类表面活性剂的实例包括蔗糖脂肪酸盐/酯、脱水山梨糖醇脂肪酸酯、聚氧化乙烯脱水山梨糖醇脂肪酸酯、聚氧化乙烯脂肪酸酯和烷醇酰胺。非离子表面活性剂优选具有700~2000的分子量。当该分子量低时,悬浮在空气中的硅氧烷具有较高分子量且因此用具有较高分子量的硅氧烷置换所述表面活性剂的可能性增加。当分子量高时,所述表面活性剂不太可能结合到化合物半导体衬底的表面且因此不太可能形成具有期望厚度的膜。因此,最佳分子量为800~2000。表面活性剂溶液的ph优选在弱酸性范围内。当所述溶液为强酸性时,在处理期间粒子倾向于附着到表面上。因此,所述ph优选为2~7。所述ph可以用无机酸如盐酸或硝酸,或者有机酸如柠檬酸、苹果酸或乙酸调节。在表面活性剂中进行浸渍的时间不受特别限制,只要其为10秒以上即可。然而,长时间浸渍造成经济不利。当时间小于10秒时,所形成的表面活性剂层不具有足够大的厚度。在550℃的生长温度下在根据本发明处理的化合物半导体衬底上生长外延膜,而在该生长之前不进行高温热清洁。在外延膜与化合物半导体衬底之间的界面处通过sims分析测定的硅(si)峰值浓度为2×1017原子/cm3以下。[实施例1]准备gaas衬底。各gaas衬底通过垂直布里奇曼(vb)法(垂直船法)制造;碳掺杂量为1×1015~2×1016/cm3;晶体的氧含量为5×1016~5×1017/cm3;晶体缺陷蚀坑密度(epd)为1000/cm2以下;所述衬底为具有1×107ωcm~6×108ωcm的电阻率的半绝缘衬底;且直径为4英寸。至于衬底的面取向,使用通常用于电子器件应用中的mocvd外延的(100)2°-偏离衬底(面取向公差:±0.5°)。根据mocvd条件和器件性能将偏离(100)取向的衬底如(100)±0.5°衬底和(100)0.05°~2°-偏离衬底用作用于mocvd的衬底;且根据本发明的利用非离子表面活性剂的处理可以对所述衬底中的任一种提供类似的优点。当晶体为多晶体时,杂质沿晶粒间界集中。当在这种多晶体上形成器件时,电流可能流过晶粒间界。因此,晶体需要是单晶体。当衬底具有小于1×107ωcm的电阻率时,在施加电压时,电流漏出具有约1μm厚度的高纯度gaas层而到达衬底。因此,在用于hemt的外延衬底中,衬底的电阻率同样重要且必须为1×107ωcm以上。另一方面,虽然异质结双极晶体管(hbt)为gaas高速器件的一个实例,但在半绝缘衬底上形成具有约1×1018原子/cm3的浓度的n-型集电器层。因此,即使在衬底界面处存在约1×1018原子/cm3的杂质,也不会造成问题。将gaas衬底的表面镜面抛光且然后对所述衬底进行标准清洁。参照图4,通过使用分批清洁设备,随后将衬底在具有700~900分子量的非离子表面活性剂a、具有800~1200分子量的非离子表面活性剂b和具有800~1000分子量的非离子表面活性剂c(聚氧化烯烷基醚)的0.1~3体积%水溶液中浸渍5~120秒(处理时间),然后用超纯水适当冲洗预定的时间(冲洗时间(秒)),且随后进行旋转干燥。将已经浸渍在表面活性剂中的衬底和未浸渍在表面活性剂中的衬底置于标准晶片托盘中,进行氮气吹扫,且然后密封并通过相同的储存方法在相同的储存场所储存在氧气和水分不能透过的清洁袋中。所使用的晶片托盘和袋不含有机si化合物(硅氧烷)(硅氧烷释气量小于0.01μg/g)。以如下方式进行晶片托盘和袋的释气分析:将晶片托盘和袋精细地切割且然后加热到40℃,并用气相色谱-质谱联用仪(gc-ms)分析所产生的物质。在此进行的标准清洁为在镜面抛光表面之后进行的一般清洁方法。具体地,采用以下方法:(1)在室温(25℃)下在氢氧化四甲铵的0.5体积%水溶液中浸渍5分钟(2)超纯水冲洗3分钟(3)在室温(25℃)下在氢氧化四甲铵的0.5体积%水溶液中浸渍5分钟(4)超纯水冲洗3分钟(5)在室温(25℃)下在具有ph5的硝酸酸性水溶液中浸渍3分钟(6)超纯水冲洗3分钟(两次)(7)旋转干燥或者,可以使用通常已知的其他清洁方法。在干燥之后即刻测定表面活性剂层的厚度。表面活性剂层的厚度可以用椭圆偏光仪(rudolphautoeliv)测定。至于椭圆偏光仪的参数,测定光波长为632.8nm;衬底折射率为3.857;衬底消光系数为0.217;为了方便计算,将表面活性剂层的折射率固定为1.8。在具有表面活性剂层的衬底表面内,衬底的天然氧化膜层在表面活性层下面。天然氧化膜层具有约0.7nm的厚度。从精度方面来说,椭圆偏光仪很难通过测定和分析而单独确定天然氧化膜的小厚度和在天然氧化膜下面的表面活性剂层的小厚度。因此,在本发明中,将包括天然氧化膜和表面活性剂层两者的膜(层)的厚度确定为“膜厚度”。在不使用表面活性剂的情况下,表面活性剂层的膜厚度指示天然氧化膜的厚度。在储存20天之后,将各衬底从密封袋中取出且通过mocvd在衬底上形成外延膜。利用通用的mocvd反应器进行该外延生长而在生长之前不进行高温热清洁以在550℃的生长温度下形成未掺杂的1-μmgaas外延膜。然后测定各衬底的方块电阻。另外,利用sims测定在未掺杂的外延膜(epi-膜)和衬底之间的界面处的si含量。将结果描述在表i中。[表i]如表i中所指示的,在浸渍在表面活性剂中的衬底之中,其中表面活性剂层的厚度超过1.50nm的衬底具有10.0×103ω/□以上的高方块电阻。未浸渍在表面活性剂中的现有产品具有5.6×103ω/□以下的低方块电阻。利用sims测定在外延膜与衬底之间的界面处的si含量。结果,在浸渍在表面活性剂中的衬底之中,其中作为膜厚度的表面活性剂层的厚度超过1.50nm的衬底具有2.0×1017原子/cm3以下的峰值浓度,其污染非常少;未浸渍在表面活性剂中的现有产品中的一些具有显著超过5×1017原子/cm3的浓度。因此,存在明显的差别。当作为膜厚度的表面活性剂层的厚度小于1.5nm时,在外延膜与衬底之间的界面处的si含量相对于现有产品没有显著减小且方块电阻没有变得非常高;因此没有充分提供表面活性剂层的效果。另一方面,当膜厚度为1.5nm以上时,方块电阻变得足够高。然而,当膜厚度超过3.0nm时,外延生长之前的衬底的表面具有白色混浊性或所得外延膜具有表面不规则,因为在加热到外延生长温度期间厚的表面活性剂层不太可能蒸发。因此,作为膜厚度的表面活性剂层的厚度优选为1.5nm以上且3.0nm以下;特别优选1.5nm~2.0nm,因为si污染低且归因于由外延生长期间膜的不充分分解(蒸发)引起的界面处的碳和氧的残留的污染的可能性非常低。表面活性剂层的厚度可以用椭圆偏光仪容易地测定,且基于该厚度,可以推定在外延生长之后界面处的si含量。因此,可以更确实地进行衬底的品质控制。当对化合物半导体衬底进行标准清洁时,在清洁之后即刻衬底表面上的天然氧化膜保持不稳定;在过去约一周之后,氧化膜变得稳定,且在此期间,衬底表面上的膜厚度增加约0.3nm。在其中如本发明中那样形成表面活性剂层的衬底中,相对于涂布表面活性剂之后即刻的膜厚度,在过去一周之后,膜厚度也增加约0.3nm。对以与表i中的16、17、19、20、27和28号相同的方式清洁并储存的其他衬底的表面进行tof-sims分析。对于各种情况,对两个衬底进行分析。tof-sims分析对具有表ii中列出的质量数的阳离子进行检测。将所检测的离子的计数描述在表ii中。在本发明的产品中,将衬底的表面用图5中例示的表面活性剂覆盖。然而,在通过tof-sims分析得到的结果中,没有检测到图5中所示的具有高分子量的原始离子,并且检测到了由c、h和o构成的多个有机离子。这可能是因为在测定时作为一次离子辐射的69ga+部分地切断表面上的表面活性剂的键。将相对强度描述在表iii中,所述相对强度通过限定在1~1000的质量数范围内检测到并也包含具有低计数且未描述在表ii中的离子物种的离子物种的总计数作为分母并通过限定目标离子的计数作为分子来确定。[表ii]质量式27号16号19号17号24mg+13925233640ca+189817128963cu+14289222915ch3+778817313141141562927c2h3+2240943735489225004241c3h5+2350240250793004266477c6h5+1277556506453191c7h7+10745172907568219c15h23o+717613534931ch3o+408615621201631236745c2h5o+519254394845455990659c3h7o+23451688648663313573c3h5o2+260353413107350087c4h7o2+882342113792338101c5h9o2+1275533719192115c6h11o2+1663572474724387387+6258010457457+720876545545+317816603603+010413607607+19412633633+11757418nh4+2443020511272212144c2h6n+42171198606118746c2h8n+82141977339110360c3h10n+1163424229112575as+1605815248635916879160gaaso+1527101586893177gaaso2h+1036848106675245ga2aso2+26719524156261ga2aso3+20622015137283gaas2o4+349223332907171ga+111570610759315219371158196总计总计+3314251339579224429243800407[表iii]基于tof-sims分析的结果,将在由c、h和o构成的有机离子的质量数下检测的相对强度与表面si含量(在epi-膜/衬底界面处的si峰值浓度)之间的关系示出在图6和图7中。表iii指示在本发明的产品中含有h、c和o的有机物质的检测量大。例如,根据图6,当在31的质量数下的相对信号强度为2.4×10-3以上时,表面中的si峰值浓度变成2×1017以下;根据图7,当在73的质量数下的相对信号强度为3.2×10-4以上时,表面中的si峰值浓度变成2×1017以下。在本发明的产品与标准清洁产品之间的比较指示,当在31的质量数下的相对信号强度为标准清洁产品(27号)的相对信号强度的2.0倍以上时,si峰值浓度变成2×1017以下,且对于在73的质量数下的信号,其为4.1倍以上。在标准清洁的情况下,表面未用长链c覆盖,且因此不具有抗硅氧烷性;通过tof-sims测定的在诸如31和73的质量数下的相对信号强度基本为类似的低值;且界面si含量响应气氛中硅氧烷的变化而迅速改变。另外,对以与16、17和27号(标准清洁产品)相同的方式清洁并储存的其他衬底进行同步辐射xps分析。以如下方式进行分析:将九州同步光研究中心中的光束线(bl12)用作光源,入射x射线能量为365ev,通能为11.75ev且发射电子飞离角为90°。将xps光谱集体示出在图8中。本发明的产品在比在285ev下的c1s的峰高1~2ev的能量下具有高能量侧的峰。衬底的si含量越低,高能量侧的峰变得越高。根据光谱,对在285ev下的峰的积分强度与高能量侧的峰的积分强度进行比较。因此,图9指示,为了使si峰值浓度为2×1017以下,高能量侧的峰的积分强度为在285ev下的峰的积分强度的0.25倍以上。[实施例2]准备gaas衬底。各gaas衬底通过垂直梯度冷凝(vgf)法(垂直船法)制造;碳掺杂量为1×1015~2×1016/cm3;晶体的氧含量为5×1015~5×1016/cm3;晶体缺陷epd(蚀坑密度)为500/cm2以下;所述衬底为具有1×107~1×108ωcm的电阻率的半绝缘衬底;且直径为6英寸。至于衬底的面取向,使用(100)2°-偏离衬底(面取向公差:±0.5°)。将gaas衬底的表面镜面抛光。然后如实施例1中那样使用标准清洁溶液对衬底进行分批清洁并暂时干燥。然后,用单晶片加工设备对各衬底进行加工:将衬底利用通过稀释具有800~1000分子量的非离子表面活性剂(聚氧化烯烷基醚)的30体积%水溶液而制备的0.35~5体积%溶液,在200~1000rpm的转速下旋转涂布10~60秒,且然后在2000rpm的高转速下干燥35秒。在单晶片涂布的情况下,利用非离子表面活性剂的水溶液对前表面进行涂布;当水溶液到达后表面时,可能造成后表面的混浊性。因此,将后表面用超纯水冲洗。另外,在干燥时,为了促进后表面的干燥,将氮气喷雾到后表面以促进干燥。此后,如实施例1中那样利用椭圆偏光仪测定表面活性剂层的厚度。将衬底置于干净的pp(聚丙烯)晶片托盘中,进行氮气吹扫,且然后密封,并通过与实施例1中相同的储存方法在相同的储存场所将其储存在氧气和水分不能透过的清洁袋中。在储存20天之后,从密封袋中取出各衬底且如实施例1中那样通过mocvd在衬底上形成未掺杂的1-μmgaas外延膜。在形成外延膜之后,测定各衬底的方块电阻。另外,利用sims测定在外延膜与衬底之间的界面处的si含量。将结果描述在表iv中。[表iv]表iv指示,即使在用表面活性剂进行单晶片涂布的情况下,方块电阻也为10×103ω/□以上的高值且在界面处的si峰值浓度也为2×1017原子/cm3以下。在清洁之后即刻的表面活性剂层的膜厚度在一个有效数字下为1.5nm~3.0nm。[实施例3]准备gaas衬底。各gaas衬底通过vgf法(垂直船法)制造;碳掺杂量为1×1015~2×1016/cm3;所述衬底为具有1×107~5×108ωcm的电阻率的半绝缘衬底;且直径为6英寸。至于衬底的面取向,使用(100)2°-偏离衬底(面取向公差:±0.3°)。对gaas衬底的表面进行加工而使其变成镜面。然后,使用标准清洁溶液对衬底进行分批清洁,且在干燥时,进行滴干。具体地,根据以下流程进行处理:晶片清洁溶液(有机碱性溶液)→超纯水冲洗→晶片清洁溶液→超纯水冲洗→酸清洁→超纯水冲洗→超纯水冲洗→滴干(表面活性剂处理)。如实施例1中那样进行利用标准清洁溶液的清洁,不同之处在于,有机碱性溶液为氢氧化四甲基铵且用0.1%盐酸进行酸清洁。以如下方式进行滴干。将具有800~1100分子量的非离子表面活性剂(聚氧化乙烯壬基苯基醚)的30体积%水溶液稀释成0.5%溶液并使用。将干燥器的上部气氛用高纯度氮气吹扫。将化合物半导体衬底在含有上述水溶液的干燥槽中浸渍2分钟且然后以恒定速率缓慢取出并干燥。具体地,在取出干燥时,在化合物半导体衬底的表面上形成非离子表面活性剂的膜且非离子表面活性剂的亲水性基团(形成氢键的部分)与衬底表面形成氢键;因此,将非离子表面活性剂的疏水性基团布置在衬底表面上且因此衬底表面展示疏水性。因此,一将衬底从水溶液中取出而进入氮气氛中,水就从衬底表面滴下。因此,通过连续缓慢提起衬底,在衬底表面上形成表面活性剂层,同时干燥衬底表面。作为用椭圆偏光仪测定的膜厚度,在这样处理的衬底表面上的表面活性剂层具有2.0nm的厚度。将衬底置于干净的pp(聚丙烯)晶片托盘中,进行氮气吹扫,且然后密封,并通过与实施例1中相同的储存方法在相同的储存场所将其储存在氧气和水分不能透过的清洁袋中。在储存20天之后,将各衬底从密封袋中取出且如实施例1中那样通过mocvd在衬底上形成外延膜。在形成外延膜之后,测定衬底的方块电阻。结果,方块电阻为9.9×104ω/□的高值。利用sims测定在外延膜与衬底之间的界面处的si峰值浓度且发现在该界面处的si峰值浓度为1.1×1017原子/cm3。[实施例4]准备gaas衬底。各gaas衬底通过vb法(垂直船法)制造;如实施例2中那样将衬底用碳掺杂;晶体的氧含量为1×1016~1×1017/cm3;晶体缺陷epd(蚀坑密度)为1000/cm2以下;所述衬底为具有0.5×108~2.0×108ωcm的电阻率的半绝缘衬底;且直径为6英寸。至于衬底的面取向,使用(100)2°-偏离衬底(面取向公差:±0.5°)。从铸块切出gaas衬底。实施衬底的外周面倒角。然后,用硬质抛光布对衬底进行双侧抛光,使得衬底的前表面和后表面两者都变成镜面。用硬质抛光布对在其上要形成器件的表面进一步进行镜面精抛光,使得表面具有0.10nm的表面粗糙度rms和1.5μm的平坦度总厚度变化(ttv)。然后,如实施例1中那样使用标准清洁溶液对衬底进行分批清洁且暂时干燥。在标准清洁之后的48小时内,通过在1000rpm的转速下旋转涂布15秒而将表面活性剂c的0.087重量%水溶液涂布到各gaas衬底的表面,且然后在2000rpm的高转速下干燥35秒。如实施例1中那样用椭圆偏光仪测定表面活性剂层的厚度。此时,所使用的表面活性剂的未稀释溶液为以使得除c、h和o以外的杂质的量为50ng/mm3以下的方式提纯并控制的溶液。用水平保持晶片(衬底)的单晶片旋转处理设备进行利用表面活性剂的处理;且将其中进行处理的气氛的si浓度控制为小于0.5μg/m3。具体地,将用于除去气氛中的含硅有机物质(硅氧烷)的活性炭过滤器连接到空调系统;将未被si污染且由氟烃树脂形成的超高效空气(ulpa)过滤器连接到其中进行涂布的处理室中;因此,尘埃浓度小于1粒子/ft3。利用安置在外周的三个点处的聚酰亚胺树脂插脚支撑晶片(衬底)而使得来自外周部分的污染最小化。在进行表面活性剂处理的同时,将超纯水供给到后表面以防止表面活性剂到达后表面。在干燥期间,从后表面喷嘴喷出氮气以加速后表面的干燥。结果,获得具有膜的衬底,其中膜具有表v中的膜厚度;表面上的膜厚度变化小于±0.3nm;在表面上具有0.3μm以上的尺寸的外来粒子的数目为50个以下(除了距外周3mm内的区域之外的晶片整个表面,该区域充当测定装置用边缘部分);且用全反射x射线荧光(txrf)设备分析的表面上的重金属的量小于10×1010原子·cm2。当将表面通过标准清洁处理且然后长时间静置时,进行表面的氧化且结果,发生从清洁之后即刻的亲水性表面到疏水性表面的表面状态转变。因此,表面活性剂变得不太可能附着到表面且变得不太可能在表面上形成膜。另外,进行气氛中的si到表面的附着。因此,表面的si含量没有充分减小。因此,理想地在48小时内进行处理,如果有可能,则在进行标准清洁之后的24小时内进行处理。利用由tencorcorporation制造的surfscan6220测定表面上的外来粒子的数目。因为表面上的外来粒子在形成外延膜之后变成缺陷,所以期望使其数目最小化。因为表面上的重金属在形成外延膜之前的加热时不太可能蒸发,所以大量的重金属造成界面处的渗漏。因此,重金属的量期望为至少50×1010原子/cm2以下,如果有可能,则为10×1010原子/cm2以下。将衬底置于干净的pp(聚丙烯)晶片托盘中,进行氮气吹扫,且然后将其密封并储存在氧气和水分不能透过的清洁袋中。至于储存容器,使用如实施例1中那样满足小于0.01μg/g的硅氧烷释气量且总释气量小于0.5μg/g的托盘。当释气量大时,造成表面污染,这可能造成膜厚度变化。如实施例1中那样通过mocvd在改变了在表面活性剂处理之后的储存时间的衬底上形成外延膜。对于各衬底,测定在外延膜与衬底之间的界面处的si含量和方块电阻。将结果描述在表v中。仅进行标准清洁的现有情况具有如下趋势:储存时间越久,界面si含量越高且方块电阻越低。相反,在进一步进行表面活性剂处理的情况下,即使储存时间变成90天的长时间,也将界面si含量抑制为低值且将方块电阻保持在高于测定装置的测定极限的高电阻下。[表v]根据本发明,包含si的杂质对衬底表面的附着量小,在杂质蒸发时造成的表面粗糙化受到抑制且界面结晶性和平坦性的劣化受到抑制,这可能导致器件特性增强。因此,根据本发明,即使进行低温清洁并形成外延膜时,也可以减小漏电流。因此,通过减少制造时间并减少充当在清洁期间消耗的原料气体的胂的消耗量,提供成本降低的重大优势。表v指示,在本发明中在外延/衬底界面处o和c的量也小。当由n型衬底制造器件使得电流从外延表面流动到衬底的后表面时,可以抑制在界面处载流子的减少且可以抑制操作电压的升高。另外,当由半绝缘衬底制造电子器件时,可以抑制背栅效应和侧栅效应且可以改善高频特性。因此,除了减小界面si含量的效果之外,还期待通过减少其他杂质而提供的效果。[产业实用性]根据本发明,可以提供半绝缘的化合物半导体衬底,其中,即使在长时间储存之后,也不太可能出现电特性异常且可以在低成本下实现hemt的外延生长。[附图标记]1:半绝缘gaas衬底2:i-gaas层(高纯度外延层)3:n-algaas层(电子供给层)4:源电极5:栅电极6:漏电极11:正常电流流动12:异常电流流动20:表面活性剂21:有机污染当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1