一种杂原子改性氟化碳量子及其制备方法与流程
本发明属于新型氟化碳材料制备及其电池应用领域,具体涉及一种杂原子掺杂氟化碳量子点材料及其制备方法。
背景技术:
传统氟化碳是一种由碳、氟两种元素组成的共价型层间化合物,其化学式为(cfx)n。其传统制备方法为由石墨与氟气或含氟物质进行氟化反应,制备得到氟化石墨。氟化碳材料可以作为锂一次电池的正极材料,与金属锂负极组合成为锂氟化碳电池。锂氟化碳电池具有一次电池最高的理论比能量约2180wh/kg。传统方法制备的氟化碳材料即氟化石墨材料,由于材料的颗粒尺寸和孔径结构等受限域氟化前驱体石墨的影响和制约,氟化碳材料放电很难达到其理论比容量。其次,由于石墨晶格完整,较难进行氟化,制备的氟化碳材料的结构很难突破其氟碳比等于1的限制。f与c的键合方式可以分为共价键和离子键,通常在炭前驱体的晶格缺陷位置以及其结构不均一位点处,可以形成更多的f-c键或者获得更多离子态的f-c键,因此我们通过对氟化前驱体的结构设计,经过精准可控氟化技术,即采用研发制备的杂原子参杂炭量子点为氟化前驱体,利用气相氟化技术,获得更高f/c以及含有更多离子态f-c键的氟化碳量子点材料。利用这种工艺有效提高氟化碳材料的放电比容量和比能量。通过电池工艺的开发制备出具有高比能量的氟化碳电池。
氟化碳材料的结构和性质,极大地影响锂氟化碳电池的电化学性能。原材料的结构和性质,如石墨化程度、材料的维数等,以及氟化工艺是影响氟化碳性能的关键因素。近年来,对氟化碳在电池领域的应用研究主要集中在材料改性方面,如通过控制氟化的条件、采用表面包覆技术以及控制热裂解等技术,在不改变氟化碳材料自身结构的前提下提高碳的含量,提高材料的导电性,进而提高电池的功率密度。但是,上述改性并未显著改变氟化碳材料的结构,不能提高锂离子在氟化碳材料层间的固相扩散速率,因此对氟化碳材料的放电倍率性能的提高帮助有限。
对氟化的前驱体进行结构设计,可以有效调控制备的氟化碳材料的结构,进而调控氟化碳材料的电化学性能。当炭的结构维度减小到零维或准零维尺寸时,炭材料具有更多的边缘不饱和位置,在氟化过程中可以与更多的f进行化学键合,形成更多的f-c键,因而可以有效提高f/c,获得高比容量的氟化碳材料。
技术实现要素:
本发明的目的在于提供一种杂原子掺杂氟化碳量子点及其制备方法。
为了达到上述目的,本发明采用如下技术方案:采用研制的5~20nm的杂原子掺杂炭量子点材料为氟化前驱体,利用含氟化合物对其进行一定温度和压力下的氟化处理,样品经纯化后处理,得到杂原子改性氟化碳量子点材料。
一种杂原子掺杂氟化碳量子点及其制备方法,该制备方法步骤如下:采用有机小分子单体为反应前驱体,利用可控的溶液化学法,结合微波辅助作用,经过石墨化处理,制备高结晶度的碳量子点。再经过可控的氟化技术,制备杂原子改性的氟化碳量子点。
制备方法中,所使用的有机小分子物质为葡萄糖、柠檬酸、聚亚苯基前驱体、苯胺、吡咯、乙烯基苯硼酸等中的一种或几种混合物。有机小分子反应物的质量百分浓度为10~0.5mg/ml。
制备方法中,溶液化学法所采用的反应体系溶剂为乙醇和水的混合溶液,水:乙醇=1:1~5。在溶液化学反应体系中添加磷酸、硼酸或三氯化铁等中的一种或两种,添加量与有机小分子反应物的质量比为1:100~200。
制备方法中,反应体系中采用微波辅助聚合,微波功率为50~600w,辅助合成为间歇性辅助合成,每次微波时间为3min,间隔时间为5min,总合成反应时间为1~6h。
制备方法中,石墨化处理的温度为600~1200℃,石墨化处理时间为2~4h。
制备方法中,对碳量子点的氟化采用的氟/氮混合气的氟气浓度为5%~20%,氟化处理时间为1h~4h,氟化温度为200~400℃。
与现有技术相比,本发明的有益效果:
1.制备氟化碳材料结构上具有5-20nm的小的颗粒尺寸,电化学性能方面具有优良的倍率性能。
2.由于氟化前驱体的小尺寸化,导致炭结构的不饱和度增加,从而氟化时,可以嫁接更多的f,因而材料具有高的f/c,可以达到1.2。
3.材料具有高的放电比容量,0.1c放电比容量高达985mahg-1,材料能量密度高达2300wh/kg。
附图说明
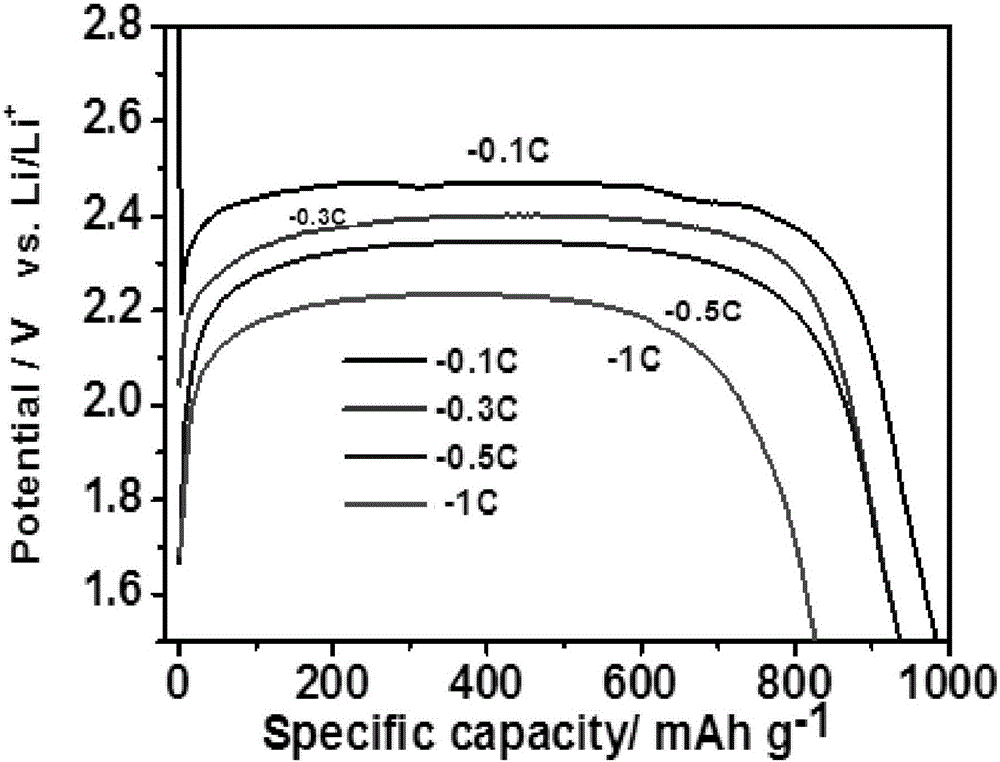
图1为实施例1所得材料的电化学性能图。
具体实施方式
下面的实施例可以使本领域的普通技术人员更全面地理解本发明,但不以任何形式限制本发明。
实施例1
取1.0g葡萄糖,溶解于100ml去离子水和200ml无水乙醇的混合溶液中,搅拌均匀,加入0.01g硼酸,搅拌至溶解。将反应体系放入微波辅助合成反应器中,微波辅助聚合,微波功率为600w,总反应时间6h。反应后样品进行过滤,60℃干燥处理。然后在氮气保护气条件下,600℃炭化2h,再1200v1h制备得到含硼炭量子点材料。将制备的炭材料在50mlmin-1f2流量下,进行200℃氟化处理4h,得到硼掺杂氟化炭量子点材料。制备的材料的氟含量为58wt%,杂原子硼的含量为2wt.%;f/c摩尔比为0.96。0.1c1.5v截止电压放电比容量为985mahg-1,放电电压为2.40v,1c1.5v截止电压的放电比容量为810mahg-1,放电电压为2.15v。电化学性能如图1所示。
实施例2
取1.0g苯胺,溶解于100ml去离子水和200ml无水乙醇的混合溶液中,搅拌均匀,加入0.01g三氯化铁,加入0.005ml的磷酸,搅拌至溶解。将反应体系放入微波辅助合成反应器中,微波辅助聚合,微波功率为500w,总反应时间5h。反应后样品进行过滤,60℃干燥处理。然后在氮气保护气条件下,600℃炭化2h,再1200℃1h制备得到含氮炭量子点材料。将制备的炭材料在10mlmin-1f2流量下,进行250℃氟化处理4h,得到氮磷掺杂氟化炭量子点材料。制备的材料氟的质量百分含量为55wt.%,杂原子氮的质量百分含量为1.5wt.%,杂原子磷的质量百分含量为3wt%;f/c摩尔比为0.89。0.1c1.5v截止电压放电比容量为920mahg-1,放电电压为2.42v,1c1.5v截止电压的放电比容量为810mahg-1,放电电压为2.10v。
实施例3
取1.0g吡咯,溶解于100ml去离子水和200ml无水乙醇的混合溶液中,搅拌均匀,加入0.01g三氯化铁,加入0.005ml的硼酸,搅拌至溶解。将反应体系放入微波辅助合成反应器中,微波辅助聚合,微波功率为300w,总反应时间4h。反应后样品进行过滤,60℃干燥处理。然后在氮气保护气条件下,600℃炭化2h,再1200℃1h制备得到含氮炭量子点材料。将制备的炭材料在20mlmin-1f2流量下,进行400℃氟化处理4h,得到氮硼掺杂氟化炭量子点材料。制备的材料氟的质量百分含量为60wt.%,氮的质量百分含量为1.5wt.%,硼的质量百分含量为2wt.%;f/c摩尔比为1.12。0.1c1.5v截止电压放电比容量为920mahg-1,放电电压为2.42v,1c1.5v截止电压的放电比容量为810mahg-1,放电电压为2.10v。
实施例4
取1.0g乙烯基苯硼酸,溶解于100ml去离子水和250ml无水乙醇的混合溶液中,搅拌均匀,加入0.005g三氯化铁,加入0.01ml的磷酸,搅拌至溶解。将反应体系放入微波辅助合成反应器中,微波辅助聚合,微波功率为400w,总反应时间6h。反应后样品进行过滤,60℃干燥处理。然后在氮气保护气条件下,600℃炭化2h,再1100℃1h制备得到含硼磷炭量子点材料。将制备的炭材料在10mlmin-1f2流量下,进行300℃氟化处理4h,得到硼磷掺杂氟化炭量子点材料。制备的材料的氟含量为54wt.%,杂原子磷的含量为3wt.%,硼的含量为1wt%;f/c摩尔比为。0.1c1.5v截止电压放电比容量为905mahg-1,放电电压为2.42v;1c1.5v截止电压的放电比容量为830mahg-1,放电电压为2.13v。
实施例5
取1.0g葡萄糖,溶解于150ml去离子水和250ml无水乙醇的混合溶液中,搅拌均匀,加入0.01g硼酸,加入0.005ml的磷酸,搅拌至溶解。将反应体系放入微波辅助合成反应器中,微波助聚合,微波功率为400w,总反应时间6h。反应后样品进行过滤,60℃干燥处理。然后在氮气保护气条件下,600℃炭化4h,再1100℃1h制备得到含硼磷炭量子点材料。将制备的炭材料在10mlmin-1f2流量下,进行300℃氟化处理4h,得到硼磷掺杂氟化炭量子点材料。制备的材料的氟含量为55wt.%,杂原子磷的含量为1wt.%,硼的含量为1.5wt%;f/c=0.91。0.1c1.5v截止电压放电比容量为905mahg-1,放电电压为2.42v;1c1.5v截止电压的放电比容量为830mahg-1,放电电压为2.13v。
- 还没有人留言评论。精彩留言会获得点赞!