高分散负载型核壳结构Pd@Ni/WC直接醇类燃料电池催化剂及其制备方法与流程
本发明属于能源材料及电化学技术领域,涉及一种直接醇类燃料电池阳极电催化剂,具体涉及到一种核壳结构pd@ni/wc催化剂及其制备方法。
背景技术
燃料电池是将燃料的化学能直接转化为电能的高效洁净发电装置,对于解决当今世界面临的“能源短缺”和“环境污染”这两大难题具有重要的意义,被认为是21世纪最为重要的能源动力之一。燃料电池以其理论能量密度高、无毒、洁净、存储和运输方便、安全等优点,在近几年获得了广泛的关注,具有十分广阔的应用前景。
直接醇类燃料电池是使用小分子醇,如ch3oh、c2h5oh、(ch2oh)2等作为燃料的燃料电池,具有燃料来源丰富、易于储存并且携带方便等优势,是国内外研究的热点。然而,如何提高阳极催化剂的活性与稳定性仍直接制约着直接醇类燃料电池的工业化进程。在众多阳极电催化剂中,pd/c催化剂是应用较为广泛的一种,但其成本较高且稳定性较差。研究发现通过掺杂第二金属(pd-m,m=pt、au、ir、sn、bi、co、ni)或者非金属(例如磷)可以提高pd催化剂的活性和稳定性,但传统简单混合掺杂对成本的降低值有限。另外,将金属纳米粒子负载在一些具有良好导电性及大比表面积的载体上,也可以一定程度提高金属的利用率,抑制纳米粒子的聚集进而提高催化剂的活性和稳定性,但如何选择合适的载体仍极具挑战。
文献(newj.chem.2017,41,13408-13417)采用化学还原法,用硼氢化钠还原ni2+,在vulcan碳载体的表面上形成ni纳米颗粒,然后使pt4+,pd2+或ru3+离子在ni纳米颗粒上沉积,形成核壳结构,并研究了其对直接硼氢化物-过氧化氢燃料电池性能的影响。该方法制备的核壳结构催化剂提高了pd的利用率和催化性能,但是vulcan碳载体只作为导电性良好的载体,对催化剂性能提高没有影响。
针对上述问题,本发明中采用偏钨酸铵、葡萄糖为钨、碳原料、以廉价易得sio2溶胶为模板,制备多孔的碳化钨,碳化钨具有类pt结构,可能和pd@ni之间形成协同效应,提高催化剂的催化活性,然后采用简单易行的浸渍还原法担载金属镍,制备高分散碳化钨载镍前体,最后采用置换法用氯化钯置换碳化钨载镍前体得到核壳结构pd@ni/wc催化剂。
技术实现要素:
本发明所解决的技术问题在于提供一种高分散负载型pd@ni/wc催化剂,可作为直接醇类燃料电池的阳极电催化剂。本发明还提供了制备所述高分散负载型pd@ni/wc催化剂的制备方法。首先制备碳化钨载体,使用sio2为模板,使得碳化钨载体具有孔结构,这有利于传质;其次碳化钨具有类pt的结构,可以有效的提高其催化活性;使用化学还原法和置换法制备得到催化性能优异的核壳结构pd@ni/wc催化剂,核壳结构降低了pd的使用率,并且核壳合金的协同作用可提高其催化活性。该制备方法简单,催化剂活性高,有利于规模化生产。
为了达到上述目的,本发明采用的技术方案为:
一种高分散负载型核壳结构pd@ni/wc直接醇类燃料电池催化剂,该催化剂首先以sio2为模板,采用高温煅烧法制得多孔结构的wc载体;再以nicl2·6h2o为ni源,采用化学还原法制得负载ni的ni/wc;最后,采用置换法,将ni表面的ni原子置换为pd,得到以ni纳米粒子为核、以pd为壳的催化剂,所得核壳结构纳米粒子大小均匀,直径为10-20nm,均匀分散于高导电性的wc载体表面。该催化剂原料成本低且来源丰富,制备过程简单,催化活性高,有利于规模化生产。
一种高分散负载型核壳结构pd@ni/wc直接醇类燃料电池催化剂的制备方法,包括如下步骤:
步骤一,制备高分散的碳化钨材料
1.1)将钨源、碳源按照质量比1~1:10溶于水中,得到混合溶液;混合溶液中加入sio2溶胶,80℃下加热搅拌1~5h,得前驱体溶液;
1.2)将1.1)中前驱体溶液干燥,得到前驱体固体;
1.3)将步骤1.2)中制得的前驱体固体,于惰性气体氛围下程序升温至500-1200℃,恒温热处理1~6h,冷却;
1.4)刻蚀步骤1.5)中制得的样品,抽滤、洗涤,干燥,制得高分散碳化钨材料;
步骤二,制备ni/wc前体
将nicl2·6h2o溶于乙二醇中,加入高分散碳化钨材料后进行超声分散;再加入硼氢化钠溶液,70℃下加热搅拌1~5h;抽滤、洗涤,干燥,制得高分散负载型ni/wc前体。所述的nicl2·6h2o与高分散碳化钨材料的质量比为1:1,所述的nicl2·6h2o、硼氢化钠的摩尔比为1:1~10。
步骤三,制备pd@ni/wc催化剂
3.1)取ni/wc前体加入乙二醇中,对样品进行超声分散,得到ni/wc混合液;
3.2)按pd、ni摩尔比为0.1~10:1向不断搅拌的ni/wc混合液中滴入h2pdcl4溶液,在室温下反应1~24h;抽滤、洗涤,干燥,制得高分散负载型核壳结构pd@ni/wc催化剂。
上述步骤1.1)中,所述的钨源包括偏钨酸铵、钨酸钠、磷钨酸等的一种或多种,碳源是葡萄糖、蔗糖、多巴胺等的一种或多种,所述的sio2溶胶粒径范围为10~500nm。
上述步骤1.2)中,所述的干燥方式包括普通干燥、真空干燥,干燥时间为1~96h,温度40~180℃。
上述步骤1.3)中,所述程序升温为从常温以1~20℃min-1的速率程序升温至500~600℃,再以1~10℃min-1的速率程序升温至700~1200℃,煅烧时间为1~6h。
上述步骤1.4)中,所述刻蚀过程可用0.1~10moll-1的氢氧化钠溶液或1~30wt.%hf溶液,所述干燥方式包括普通干燥、真空干燥,干燥时间为1~24h,干燥温度40~180℃。
上述步骤二中,所述的干燥方式包括普通干燥、真空干燥,干燥时间为1~24h,干燥温度40~180℃。
上述步骤3.2)中,所述的干燥方式包括普通干燥、真空干燥,干燥时间为1~24h,干燥温度40~180℃。
与现有技术相比,本发明所述催化剂制备方法具有以下优点:
1)采用本发明所述方法制备催化剂,由于sio2模板的隔离作用,高温烧结过程中碳化钨活性组分不易团聚,使碳化钨活性组分分散度高。
2)采用本发明所述方法制备的催化剂,试剂毒性小,安全环保,原料成本低廉,制备工艺简单,有利于大规模生产。
3)采用本发明所述方法制备的wc具有孔结构,可以为电子和物质提供了传输通道,提高了醇类的传输和电子的传导;wc较高的比表面积有利于暴露大量的金属活性位,进而有助于电化学性能的提高;而且,wc具有类pt结构,可能和pd@ni之间形成协同效应,提高催化剂的催化活性。
4)采用本发明所述方法制备催化剂,与pt基催化剂相比,pd资源丰富且成本较低,降低了燃料电池的成本。
5)采用本发明所述方法制备催化剂,由置换法制备而成,使用负载ni的前驱体,制备过程简单易行,制备过程可控,降低了pd的使用量和成本,有利于该催化剂的放大生产。
6)采用本发明所述方法制备催化剂为核壳结构。贵金属pd作为壳体分布在非贵金属ni核上不仅大大提高了pd的利用率,而且增加了pd基催化剂的活性表面积,提供更多的活性位点。另外双金属合金具有的协同结构和电子效应,可以提高其催化性能。
7)采用本发明所述方法制备催化剂,应用范围广,尤其是可以作为甲醇、乙醇和乙二醇等直接醇类燃料电池的电催化剂。
附图说明
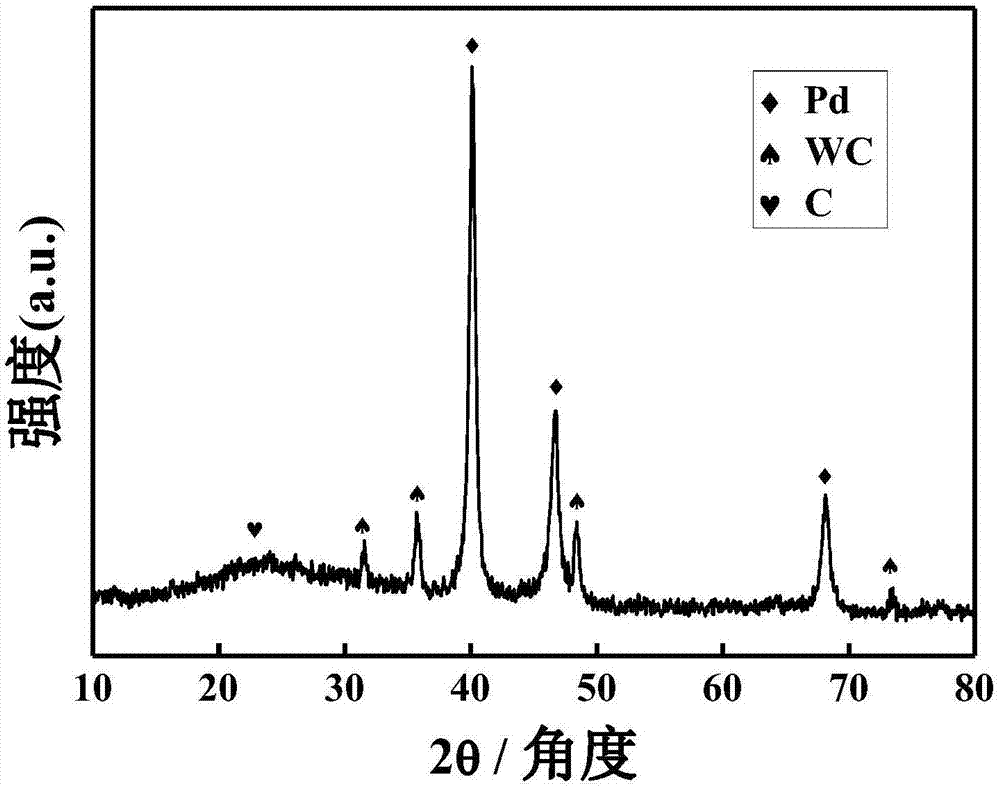
图1为根据实施例2制备得到样品pd3@ni1/wc1.31-900℃-3h的xrd谱图。
图2为根据实施例2制备得到样品pd3@ni1/wc1.31-900℃-3h的tem照片;(a)为hrtem图;(b)为tem图。
图3为根据实施例1、2、3制备得到的样品在n2饱和的1mnaoh溶液中的循环伏安曲线,扫速:20mvs-1,室温。
图4为根据实施例1、2、3制备得到的样品在n2饱和的1mnaoh+1mch3oh溶液中的循环伏安曲线,扫速:20mvs-1,室温。
图5为根据实施例1、2、3制备得到的样品在n2饱和的1mnaoh+1mch3ch2oh溶液中的循环伏安曲线,扫速:20mvs-1,室温。
图6为根据实施例1、2、3制备得到的样品在n2饱和的1mnaoh+1m(ch2oh)2溶液中的循环伏安曲线,扫速:20mvs-1,室温。
具体实施方式
下面结合实施例对本发明作详细的描述,当然本发明并不仅限于这些具体的实施例。
实施例1:pd2@ni1/wc1.31-900℃-3h(pd2@ni指pd、ni的摩尔比2:1,wc1.31表示碳化钨,钨源和碳源的质量比为1:1.31,煅烧温度900℃,煅烧时间为3h)
称取1g偏钨酸铵,1.31g葡萄糖溶于80℃去离子水中,采用naoh调节ph=11,加入2gsio2溶胶(直径20nm,质量分数40%),80℃搅拌3h;在120℃干燥5h得到前驱体固体。将上述前驱体固体在管式炉中,n2气氛下以5℃min-1的速率程序升温至550℃,然后以2℃min-1的速率程序升温至900℃,恒温反应3h,自然冷却,得前驱体复合材料。将上述前驱体复合材料置于2m的naoh溶液中,80℃搅拌12h,抽滤、洗涤,80℃真空干燥6h,制得高分散碳化钨材料wc1.31-900℃-3h。
取100mgnicl2·6h2o溶于20ml乙二醇,加入100mgwc1.31-900℃-3h,超声分散0.5h;滴加硼氢化钠溶液(0.5m,10ml),70℃搅拌1.5h,抽滤、洗涤,70℃真空干燥4h,制得目标产品ni/wc1.31-900℃-3h。
取20mgni/wc1.31-900℃-3h分散于20ml乙二醇,超声搅拌0.5h,加入氯钯酸溶液(5mm,7.04ml),搅拌12h,抽滤、洗涤,70℃真空干燥6h,制得目标产品pd2@ni1/wc1.31-900℃-3h催化剂。
实施例2:pd3@ni1/wc1.31-900℃-3h(pd3@ni指pd、ni的摩尔比3:1,wc1.31表示碳化钨,钨源和碳源的质量比为1:1.31,煅烧温度900℃,煅烧时间为3h)
称取1g偏钨酸铵,1.31g葡萄糖溶于80℃去离子水中,采用naoh调节ph=11,加入2gsio2溶胶(直径20nm,质量分数40%),80℃搅拌3h;在120℃干燥5h得到前驱体固体。将上述前驱体固体在管式炉中,n2气氛下以5℃min-1的速率程序升温至550℃,然后以2℃min-1的速率程序升温至900℃,恒温反应3h,自然冷却,得前驱体复合材料。将上述前驱体复合材料置于2m的naoh溶液中,80℃搅拌12h,抽滤、洗涤,80℃真空干燥6h,制得高分散碳化钨材料wc1.31-900℃-3h。
取100mgnicl2·6h2o溶于20ml乙二醇,加入100mgwc1.31-900℃-3h,超声分散0.5h;滴加硼氢化钠溶液(0.5m,10ml),70℃搅拌1.5h,抽滤、洗涤,70℃真空干燥4h,制得目标产品ni/wc1.31-900℃-3h。
取20mgni/wc1.31-900℃-3h分散于20ml乙二醇,超声搅拌0.5h,加入氯钯酸溶液(5mm,7.92ml),搅拌12h,抽滤、洗涤,70℃真空干燥6h,制得目标产品pd3@ni1/wc1.31-900℃-3h催化剂。
实施例3:pd4@ni1/wc1.31-900℃-3h(pd4@ni指pd、ni的摩尔比4:1,wc1.31表示碳化钨,钨源和碳源的质量比为1:1.31,煅烧温度900℃,煅烧时间为3h)
称取1g偏钨酸铵,1.31g葡萄糖溶于80℃去离子水中,采用naoh调节ph=11,加入2gsio2溶胶(直径20nm,质量分数40%),80℃搅拌3h;在120℃干燥5h得到前驱体固体。将上述前驱体固体在管式炉中,n2气氛下以5℃min-1的速率程序升温至550℃,然后以2℃min-1的速率程序升温至900℃,恒温反应3h,自然冷却,得前驱体复合材料。将上述前驱体复合材料置于2m的naoh溶液中,80℃搅拌12h,抽滤、洗涤,80℃真空干燥6h,制得高分散碳化钨材料wc1.31-900℃-3h。
取100mgnicl2·6h2o溶于20ml乙二醇,加入100mgwc1.31-900℃-3h,超声分散0.5h;滴加硼氢化钠溶液(0.5m,10ml),70℃搅拌1.5h,抽滤、洗涤,70℃真空干燥4h,制得目标产品ni/wc1.31-900℃-3h。
取20mgni/wc1.31-900℃-3h分散于20ml乙二醇,超声搅拌0.5h,加入氯钯酸溶液(5mm,8.45ml),搅拌12h,抽滤、洗涤,70℃真空干燥6h,制得目标产品pd4@ni1/wc1.31-900℃-3h催化剂。
实施例4:pd0.1@ni1/wc1-900℃-3h(pd0.1@ni1指pd、ni的摩尔比0.1:1,wc1表示碳化钨,钨源和碳源的质量比为1:1,煅烧温度900℃,煅烧时间为3h)
称取1g偏钨酸铵,1g葡萄糖溶于80℃去离子水中,采用naoh调节ph=11,加入2gsio2溶胶(直径20nm,质量分数40%),80℃搅拌3h;在120℃干燥5h得到前驱体固体。将上述前驱体固体在管式炉中,n2气氛下以5℃min-1的速率程序升温至550℃,然后以2℃min-1的速率程序升温至900℃,恒温反应3h,自然冷却,得前驱体复合材料。将上述前驱体复合材料置于2m的naoh溶液中,80℃搅拌12h,抽滤、洗涤,80℃真空干燥6h,制得高分散碳化钨材料wc1-900℃-3h。
取100mgnicl2·6h2o溶于20ml乙二醇,加入100mgwc1-900℃-3h,超声分散0.5h;滴加硼氢化钠溶液(0.5m,10ml),70℃搅拌1.5h,抽滤、洗涤,70℃真空干燥4h,制得目标产品ni/wc1-900℃-3h。
取20mgni/wc1-900℃-3h分散于20ml乙二醇,超声搅拌0.5h,加入氯钯酸溶液(5mm,0.96ml),搅拌12h,抽滤、洗涤,70℃真空干燥6h,制得目标产品pd0.1@ni1/wc1-900℃-3h催化剂。
实施例5:pd10@ni1/wc10-900℃-3h(pd10@ni1指pd、ni的摩尔比10:1,wc10表示碳化钨,钨源和碳源的质量比为1:10,煅烧温度900℃,煅烧时间为3h)
称取1g偏钨酸铵,10g葡萄糖溶于80℃去离子水中,采用naoh调节ph=11,加入2gsio2溶胶(直径20nm,质量分数40%),80℃搅拌3h;在120℃干燥5h得到前驱体固体。将上述前驱体固体在管式炉中,n2气氛下以5℃min-1的速率程序升温至550℃,然后以2℃min-1的速率程序升温至900℃,恒温反应3h,自然冷却,得前驱体复合材料。将上述前驱体复合材料置于2m的naoh溶液中,80℃搅拌12h,抽滤、洗涤,80℃真空干燥6h,制得高分散碳化钨材料wc10-900℃-3h。
取100mgnicl2·6h2o溶于20ml乙二醇,加入100mgwc10-900℃-3h,超声分散0.5h;滴加硼氢化钠溶液(0.5m,10ml),70℃搅拌1.5h,抽滤、洗涤,70℃真空干燥4h,制得目标产品ni/wc10-900℃-3h。
取20mgni/wc10-900℃-3h分散于20ml乙二醇,超声搅拌0.5h,加入氯钯酸溶液(5mm,9.68ml),搅拌12h,抽滤、洗涤,70℃真空干燥6h,制得目标产品pd10@ni1/wc10-900℃-3h催化剂。
实施例6:pd3@ni1/wc1.31-700℃-3h(pd3@ni指pd、ni的摩尔比3:1,wc1.31表示碳化钨,钨源和碳源的质量比为1:1.31,煅烧温度700℃,煅烧时间为3h)
称取1g偏钨酸铵,1.31g葡萄糖溶于80℃去离子水中,采用naoh调节ph=11,加入2gsio2溶胶(直径20nm,质量分数40%),80℃搅拌3h;在120℃干燥5h得到前驱体固体。将上述前驱体固体在管式炉中,n2气氛下以5℃min-1的速率程序升温至550℃,然后以2℃min-1的速率程序升温至700℃,恒温反应3h,自然冷却,得前驱体复合材料。将上述前驱体复合材料置于2m的naoh溶液中,80℃搅拌12h,抽滤、洗涤,80℃真空干燥6h,制得高分散碳化钨材料wc1.31-700℃-3h。
取100mgnicl2·6h2o溶于20ml乙二醇,加入100mgwc1.31-700℃-3h,超声分散0.5h;滴加硼氢化钠溶液(0.5m,10ml),70℃搅拌1.5h,抽滤、洗涤,70℃真空干燥4h,制得目标产品ni/wc1.31-700℃-3h。
取20mgni/wc1.31-700℃-3h分散于20ml乙二醇,超声搅拌0.5h,加入氯钯酸溶液(5mm,7.92ml),搅拌12h,抽滤、洗涤,70℃真空干燥6h,制得目标产品pd3@ni1/wc1.31-700℃-3h催化剂。
实施例7:pd3@ni1/wc1.31-1200℃-1h(pd3@ni指pd、ni的摩尔比3:1,wc1.31表示碳化钨,钨源和碳源的质量比为1:1.31,煅烧温度1200℃,煅烧时间为3h)
称取1g偏钨酸铵,1.31g葡萄糖溶于80℃去离子水中,采用naoh调节ph=11,加入2gsio2溶胶(直径20nm,质量分数40%),80℃搅拌3h;在120℃干燥5h得到前驱体固体。将上述前驱体固体在管式炉中,n2气氛下以5℃min-1的速率程序升温至550℃,然后以2℃min-1的速率程序升温至1200℃,恒温反应3h,自然冷却,得前驱体复合材料。将上述前驱体复合材料置于2m的naoh溶液中,80℃搅拌12h,抽滤、洗涤,80℃真空干燥6h,制得高分散碳化钨材料wc1.31-1200℃-3h。
取100mgnicl2·6h2o溶于20ml乙二醇,加入100mgwc1.31-1200℃-3h,超声分散0.5h;滴加硼氢化钠溶液(0.5m,10ml),70℃搅拌1.5h,抽滤、洗涤,70℃真空干燥4h,制得目标产品ni/wc1.31-1200℃-3h。
取20mgni/wc1.31-1200℃-3h分散于20ml乙二醇,超声搅拌0.5h,加入氯钯酸溶液(5mm,7.92ml),搅拌12h,抽滤、洗涤,70℃真空干燥6h,制得目标产品pd3@ni1/wc1.31-1200℃-3h催化剂。
实施例8:pd3@ni1/wc1.31-900℃-1h(pd2@ni指pd、ni的摩尔比3:1,wc1.31表示碳化钨,钨源和碳源的质量比为1:1.31,煅烧温度900℃,煅烧时间为1h)
称取1g偏钨酸铵,1.31g葡萄糖溶于80℃去离子水中,采用naoh调节ph=11,加入2gsio2溶胶(直径20nm,质量分数40%),80℃搅拌3h;在40℃干燥96h得到前驱体固体。将上述前驱体固体在管式炉中,n2气氛下以5℃min-1的速率程序升温至550℃,然后以2℃min-1的速率程序升温至900℃,恒温反应1h,自然冷却,得前驱体复合材料。将上述前驱体复合材料置于2m的naoh溶液中,80℃搅拌12h,抽滤、洗涤,40℃真空干燥24h,制得高分散碳化钨材料wc1.31-900℃-1h。
取100mgnicl2·6h2o溶于20ml乙二醇,加入100mgwc1.31-900℃-1h,超声分散0.5h;滴加硼氢化钠溶液(0.5m,10ml),70℃搅拌1.5h,抽滤、洗涤,40℃真空干燥24h,制得目标产品ni/wc1.31-900℃-1h。
取20mgni/wc1.31-900℃-6h分散于20ml乙二醇,超声搅拌0.5h,加入氯钯酸溶液(5mm,7.92ml),搅拌12h,抽滤、洗涤,40℃真空干燥24h,制得目标产品pd3@ni1/wc1.31-900℃-1h催化剂。
实施例9:pd3@ni1/wc1.31-900℃-6h(pd3@ni指pd、ni的摩尔比3:1,wc1.31表示碳化钨,钨源和碳源的质量比为1:1.31,煅烧温度900℃,煅烧时间为6h)
称取1g偏钨酸铵,1.31g葡萄糖溶于80℃去离子水中,采用naoh调节ph=11,加入2gsio2溶胶(直径20nm,质量分数40%),80℃搅拌5h;在180℃干燥1h得到前驱体固体。将上述前驱体固体在管式炉中,n2气氛下以5℃min-1的速率程序升温至550℃,然后以2℃min-1的速率程序升温至900℃,恒温反应6h,自然冷却,得前驱体复合材料。将上述前驱体复合材料置于2m的naoh溶液中,80℃搅拌12h,抽滤、洗涤,180℃真空干燥1h,制得高分散碳化钨材料wc1.31-900℃-6h。
取100mgnicl2·6h2o溶于20ml乙二醇,加入100mgwc1.31-900℃-6h,超声分散0.5h;滴加硼氢化钠溶液(0.5m,10ml),70℃搅拌1.5h,抽滤、洗涤,180℃真空干燥1h,制得目标产品ni/wc1.31-900℃-6h。
取20mgni/wc1.31-900℃-6h分散于20ml乙二醇,超声搅拌0.5h,加入氯钯酸溶液(5mm,7.92ml),搅拌12h,抽滤、洗涤,180℃真空干燥1h,制得目标产品pd3@ni1/wc1.31-900℃-6h催化剂。
比较例1:pd3@ni1/c(pd3@ni1指pd、ni的摩尔比3:1,c表示科琴碳kbec-600jd)
取100mgnicl2·6h2o溶于20ml乙二醇,加入100mgkb,超声分散0.5h;滴加硼氢化钠溶液(0.5m,10ml),70℃搅拌1.5h,抽滤、洗涤,70℃真空干燥4h,制得目标产品ni/c催化剂。
取20mgni/c分散于20ml乙二醇,超声搅拌0.5h,加入氯钯酸溶液(5mm,7.92ml),搅拌12h,抽滤、洗涤,70℃真空干燥6h,制得目标产品pd3@ni1/c催化剂。
比较例2:20%pd/wc1.31-900℃-3h(所述催化剂没有加入nicl2·6h2o,wc1.31表示碳化钨,钨源和碳源的质量比为1:1.31,煅烧温度900℃,煅烧时间为3h)
称取1g偏钨酸铵,1.31g葡萄糖溶于80℃去离子水中,采用naoh调节ph=11,加入2gsio2溶胶(直径20nm,质量分数40%),80℃搅拌3h;在120℃干燥5h得到前驱体固体。将上述前驱体固体在管式炉中,n2气氛下以5℃min-1的速率程序升温至550℃,然后以2℃min-1的速率程序升温至900℃,恒温反应3h,自然冷却,得前驱体复合材料。将上述前驱体复合材料置于2m的naoh溶液中,80℃搅拌12h,抽滤、洗涤,80℃真空干燥6h,制得高分散碳化钨材料wc1.31-900℃-3h。
取20mgwc1.31-900℃-3h分散于20ml乙二醇,超声分散0.5h,加入氯钯酸溶液(5mm,7.52ml);滴加硼氢化钠溶液(0.5m,8ml),搅拌12h,抽滤、洗涤,70℃真空干燥4h,制得目标产品pd/wc1.31-900℃-3h催化剂。
比较例3:20%pd/c(所述催化剂没有加入nicl2·6h2o,c表示科琴碳kbec-600jd)
取20mgkb分散于20ml乙二醇,超声分散0.5h,加入氯钯酸溶液(5mm,7.52ml);滴加硼氢化钠溶液(0.5m,8ml),搅拌12h,抽滤、洗涤,70℃真空干燥4h,制得目标产品pd/c催化剂。
图1为根据实施例2制备得到的pd3@ni1/wc1.31-900℃-3h的xrd谱图。从图1可见,衍射角2θ为40.11°、46.66°和68.12°的衍射峰分别对应的是pd(111)、(200)、(220)晶面(pcpdf#89-4897),说明实验所制备的催化剂表面含有金属钯。衍射角在2θ为31.51°、35.64°和48.3°的衍射峰分别对应的是wc(001)、(100)、(101)晶面(pcpdf#72-0097),说明实验所制备的载体含有wc。
图2为根据实施例2制备得到的样品的tem照片,图2a中粒子的晶格间距为0.225nm,归属于pd的(111)晶面,由图2b可见,金属粒子的粒径约为10-20nm,均匀分散于碳化钨载体表面。
图3为根据实施例1、2、3制备得到的样品在室温下,在n2饱和的1mnaoh溶液中的循环伏安曲线。负扫过程中0.5~0.9v(vs.rhe)所对应的峰是pdox的还原峰。
图4为根据实施例1、2、3制备得到的样品在室温下,在n2饱和的1mnaoh+1mch3oh溶液中的循环伏安曲线。可以看出各实施例的起始电位均为0.6v(vs.rhe),甲醇氧化峰电流密度大小为实施例2>实施例3>比较例2>实施例1>比较例3>比较例1。
图5为根据实施例1、2、3制备得到的样品在室温下,在n2饱和的1mnaoh+1mc2h5oh溶液中的循环伏安曲线。可以看出各实施例的起始电位均为0.45v(vs.rhe),乙醇氧化峰电流密度大小为实施例2>实施例3>比较例2>比较例3>实施例1>比较例1。
图6为根据实施例1、2、3制备得到的样品在室温下,在n2饱和的1mnaoh+1m(ch2oh)2溶液中的循环伏安曲线。可以看出各实施例的起始电位均为0.55v(vs.rhe),乙二醇氧化峰电流密度大小为实施例2>实施例3>比较例2>实施例1>比较例1>比较例3。
以上所述实施例仅表达了本发明的实施方式,但并不能因此而理解为对本发明专利的范围的限制,应当指出,对于本领域的技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些均属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!
- 磁性胶体核壳结构γ‑Fe2O3及Fe3O4的制备方法与流程
- 一种核壳结构Au@SiO2负载Pt纳米球的制备方法与流程
- 一种核壳结构Ag@TiO2@Pt纳米复合材料的制造方法与工艺
- 一种基于Ag@In核壳结构的高温钎料及其制备方法与流程
- 一种核壳结构Au@Co(OH)2纳米微球的制备方法与流程
- 一种制备核‑壳结构的高介低损钛酸铜钙基陶瓷的方法与流程
- 一种具有核壳结构的微球复合负极材料及其制备方法和应用与制造工艺
- 一种防潮耐水含橡胶籽油的核壳结构纳米改性变压器油及其制备方法与制造工艺
- 一种耐盐雾含茶籽油的核壳结构纳米改性变压器油及其制备方法与制造工艺
- 一种含锭子油的高流动性核壳结构纳米改性变压器油及其制备方法与制造工艺