琥珀酸美托洛尔缓释微丸及其制备方法与流程
本发明属于口服缓控释给药
技术领域:
,特别涉及一种含有琥珀酸美托洛尔的缓释制剂及其制备方法。更具体地,本发明涉及一种琥珀酸美托洛尔缓释微丸及其制备方法,以及包含这样的缓释微丸的片剂、胶囊剂和其他剂型。
背景技术:
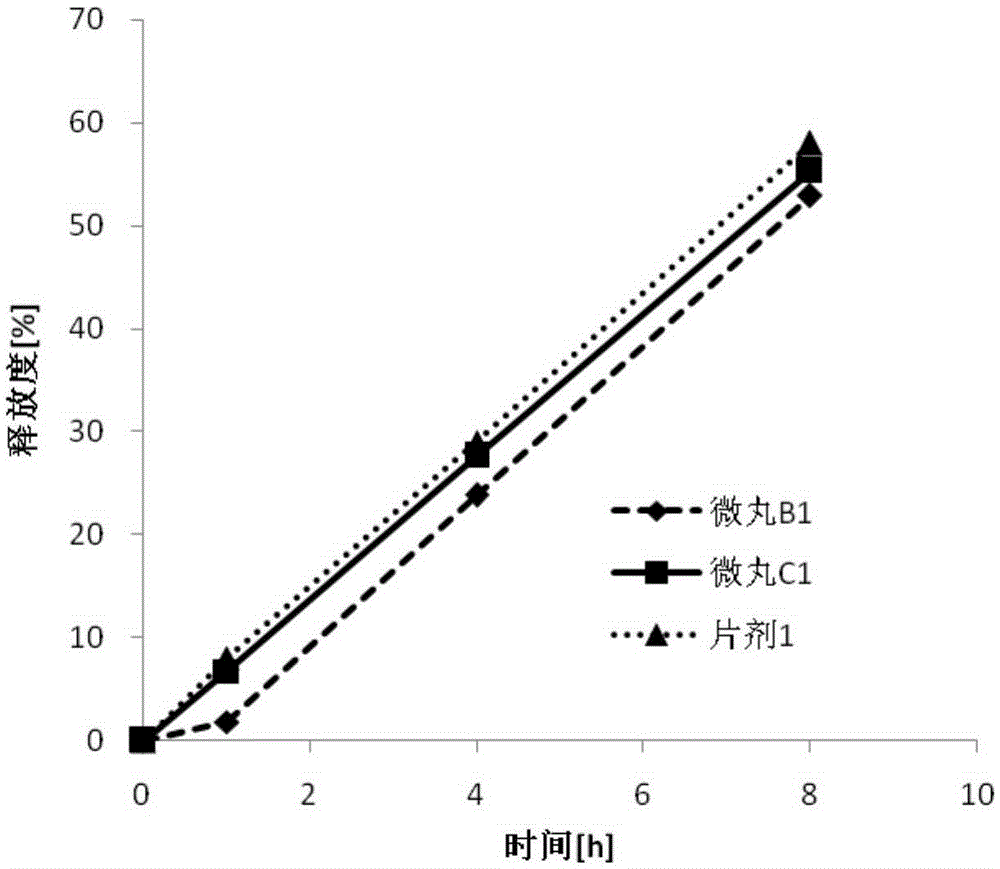
:美托洛尔是一种具有高度选择性的β1受体阻滞剂,其化学名称为1-异丙氨基-3-[对-(2-甲氧乙基)苯氧基]-2-丙醇,结构式如下:美托洛尔是治疗高血压、心力衰竭和心率失常的常用药物之一。2005年,阿斯利康公司生产的琥珀酸美托洛尔缓释片(商品名“倍他洛克”)在中国上市。目前,国内还没有同类的琥珀酸美托洛尔缓释制剂上市。琥珀酸美托洛尔缓释片是多颗粒控释单元剂型,是用琥珀酸美托洛尔缓释微丸压制成片剂,产品的释药规律具有良好的重现性和一致性,且药片可分割使用,增加了剂量的灵活性。美国专利US4927640公开了倍他洛克缓释片中的琥珀酸美托洛尔缓释微丸的制备技术。该技术以玻璃或者二氧化硅等惰性不溶性材质为起始丸芯,药物溶解在合适的溶剂中通过包衣的方法将药物涂布到起始丸芯的表面使其成为含药微丸,然后再在含药微丸表面涂布缓释层得到琥珀酸美托洛尔缓释微丸,所得的缓释微丸再和其他辅料混合后压成片剂。美国专利US5246714进一步公开了选用塑料树脂作为惰性起始丸芯制备琥珀酸美托洛尔缓释微丸的技术。但是无论是玻璃丸芯、二氧化硅丸芯还是塑料树脂丸芯,都很难在市场上买到满足药用要求的商业化产品,且这类丸芯没有统一的质量标准,难于进行质量控制。因此,该技术应用于工业生产具有一定的局限性。美国专利US20100323010公开了另外一种制备琥珀酸美托洛尔缓释制剂的技术,该技术将琥珀酸美托洛尔控释微丸和琥珀酸美托洛尔活性微丸以一定比例混合后,压制成琥珀酸美托洛尔缓释片剂。该技术在工业应用中也会面临很大的挑战,例如:该技术所用到的琥珀酸美托洛尔控释微丸和琥珀酸美托洛尔活性微丸存在粒径和密度的差异,混合均匀度存在一定风险。并且两种微丸都含有琥珀酸美托洛尔活性药物。在混合的工序中,无法通过测定混合物的含量来监控混合的均匀性。在实际生产中,两种微丸的混合均匀度难以得到保障,因此其释放很难保持批间一致性,不易于实现工业放大。中国专利CN200680054313.9公开了一种以糖丸为起始丸芯的琥珀酸美托洛尔缓释微丸的制备技术,在糖丸表面包覆一层疏水性聚合物底衣,以增强丸芯的机械强度。再进行药物层包衣、控释层包衣等工序,以得到可以用于压片的琥珀酸美托洛尔缓释微丸。该专利的实施例1揭示了初始丸芯在最终微丸中的比例会对药物的释放产生影响,当初始丸芯在最终微丸中的比例为36.6%(重量比)时,其实施例中对应的载药量为28.9%(“载药量”即活性药物在最终微丸中的重量比例)。所得制剂按照美国药典规定的释放度测定方法,在500ml,pH6.8的磷酸缓冲液介质中用桨法50rpm测定其1h的释放度为3%,而当起始丸芯在最终微丸中的比例为22.7%(重量比)时,其实施例中对应的载药量为41.5%,其1h的释放度就只有0%了。由于琥珀酸美托洛尔在胃肠道中都能被广泛吸收,因此释放延迟1h以上不利于药物迅速地发挥疗效。如果采用该专利的技术,要实现1h的释放度在3%以上,则微丸的载药量不得超过41.5%。如果最终微丸是用于压片,微丸的载药量越小,压制同等剂量规格的片剂所需要的微丸比例就越大,那么受片型大小的限制,外加辅料的比例就越小,这样的处方常常存在可压性的问题;同时由于缺少辅料的缓冲作用,这样的处方中微丸的缓释衣膜常常在压片压力的作用下遭到破坏。如果最终微丸是用于灌装胶囊,那么微丸的载药量越小,胶囊的装量就越高,胶囊的尺寸就越大,过大的胶囊尺寸不利于患者吞咽,大大的降低了患者的顺应性。此外,该专利公开的制备技术仍然需要在糖丸芯表面包覆一层疏水性聚合物底衣,以提高微丸的物理强度,增加了工艺的复杂程度。综上所述,现有的琥珀酸美托洛尔缓释微丸的制备技术均存在下述局限性中的一种或几种:a)起始丸芯没有商业化的产品,难以大批量的获得,且无统一的质量标准;b)起始丸芯在包衣之前需要先包覆一层底衣,以改善其物理性质(膨胀性、溶解性);c)需要两种不同组成的微丸以一定比例混合才能获得理想的释放曲线;d)在高载药量时(例如高于41.5%),所制备的微丸在1h内几乎没有释放。技术实现要素:本发明的目的在于提供一种可以用于压片和填充胶囊的琥珀酸美托洛尔缓释微丸及其制备方法,使其可以克服如上所述的现有技术中的诸多局限性。该琥珀酸美托洛尔制剂在按照美国药典规定的测定其释放度时,能够在1h以内释放5%以上的琥珀酸美托洛尔,并满足美国药典规定的限度要求,同时在前8h内呈现出匀速直线式的释放曲线;而且,本发明采用本领域中可商购的具有统一质量标准的丸芯,采用该技术制备的琥珀酸美托洛尔缓释微丸无需再与其他速释组分混合,不存在混合均匀性的问题,也能够保持批次间的一致性,易于实现工业放大。所制备的微丸载药量至少达到40%以上,最高可以达到60%以上,易于灌装成胶囊或压制成片剂。为实现以上目的,本发明具体如下:一种琥珀酸美托洛尔缓释微丸,其包含由内到外依次的空白丸芯、主药层、缓释层、速释药物层以及可选的缓冲层。所述琥珀酸美托洛尔缓释微丸按照美国药典方法,在500ml,pH6.8的磷酸缓冲液介质中用桨法50rpm测定其释放度,在1h以内可以释放5%以上的琥珀酸美托洛尔,并满足美国药典规定的限度要求,同时在前8h内呈现出直线式的释放曲线。所述空白丸芯为常见的商品化的市售丸芯,包括蔗糖丸芯和淀粉丸芯。所述空白丸芯优选蔗糖丸芯。根据美国药典的规定,所述蔗糖丸芯由蔗糖和淀粉组成。其中,蔗糖的比例以重量计为62.5%~91.5%之间。所述主药层由琥珀酸美托洛尔和粘合剂组成。所述粘合剂包括纤维素类衍生物、聚乙烯吡咯烷酮、聚乙二醇以及丙烯酸树脂类衍生物。所述纤维素类衍生物包括但不限于羟丙甲纤维素、羟丙纤维素、甲基纤维素、乙基纤维素。所述粘合剂优选羟丙甲纤维素、羟丙纤维素、聚乙烯吡咯烷酮。所述粘合剂在主药层中所占的比例在以重量计2%~20%的范围内。所述主药层相对于空白丸芯的增重率【增重率=100*(包衣后微丸重量-包衣前的微丸重量)/包衣前微丸重量】为360%~450%。主药层包覆空白丸芯后形成的微丸(即:包含空白丸芯和主药层的微丸)以下称作“微丸A”。所述缓释层由不溶性薄膜材料、致孔剂和可选的增塑剂组成。所述不溶性薄膜材料选自纤维素衍生物和聚甲基丙烯酸树脂的一种或几种的组合物。所述不溶性薄膜材料优选乙基纤维素。所述致孔剂选自如下水溶性聚合物中的一种或几种的组合物,包括聚乙二醇、羟丙甲纤维素、羟丙基纤维素和聚乙烯吡咯烷酮。所述致孔剂在缓释层中所占的比例范围以重量计为1%~25%,优选致孔剂在缓释层中所占的比例范围以重量计为3%~20%。所述缓释层可以可选地添加增塑剂,所述增塑剂选自聚乙二醇、柠檬酸三乙酯、癸二酸二丁酯、邻苯二甲酸二丁酯中的一种或者几种的组合物。增塑剂在缓释层中比例范围以重量计为0%~20%。所述缓释层相对于微丸A的增重率在12%~50%的范围内,优选在17%~24%的范围内。缓释层包覆微丸A后形成的微丸(即:包含空白丸芯、主药层和缓释层的微丸)以下称作“微丸B”。所述速释药物层的组成与主药层类似。所述速释药物层由琥珀酸美托洛尔和粘合剂组成。所述粘合剂包括纤维素类衍生物、聚乙烯吡咯烷酮以及丙烯酸树脂类衍生物。所述纤维素类衍生物包括但不限于羟丙甲纤维素、羟丙纤维素、甲基纤维素、乙基纤维素。所述粘合剂优选羟丙甲纤维素、羟丙纤维素、聚乙烯吡咯烷酮。所述粘合剂在速释药物层中所占的比例以重量计在2%-20%的范围内。速释层的载药量对微丸的前期释放有较大的影响,如控制不当可能使微丸呈非匀速释放,甚至形成突释或超过美国药典的限度(≤25%),在体内呈现出“双峰”式药时曲线。因此需对速释药物层的增重率进行研究,通过大量筛选和仔细优化,以得到合适的速释药物层的增重范围,使微丸前8h呈直线释放。所述速释药物层相对于微丸B的增重率在1%~20%的范围内,所述速释药物层相对于微丸B的增重率优选在3.5%~5%的范围内。速释药物层包覆微丸B后形成的微丸(即:包含空白丸芯、主药层、缓释层和速释药物层的微丸)以下称作“微丸C”。所述可选的缓冲层,可以选自低熔点的蜡质材料,所述蜡质材料选自但不限于以下材料:聚乙二醇、氢化植物油、巴西棕榈蜡、蜂蜡、石蜡、微晶蜡、卵磷脂、长链脂肪酸、长链脂肪醇。所述低熔点材料优选分子量在4000-20000之间的聚乙二醇。所述缓冲层相对于微丸C的增重率在5%~100%之间。缓冲层包覆微丸C后形成的微丸(即:包含空白丸芯、主药层、缓释层、速释药物层和缓冲层的微丸)以下称作“微丸D”。所述琥珀酸美托洛尔缓释微丸可以与其他药用辅料混合压制成片剂或填入胶囊。所得的琥珀酸美托洛尔缓释制剂在按照美国药典释放度方法测定时,1h以内可以释放5%以上的琥珀酸美托洛尔,并且能满足美国药典规定的范围要求(即1h:≤25%;4h:20%-40%;8h:60%-80%;20h:≥80%)。本发明所述的琥珀酸美托洛尔缓释制剂的制备方法如下:a)微丸A(主药层微丸)的制备:将琥珀酸美托洛尔和粘合剂溶解在适当的溶剂(比如:纯化水、丙酮、乙醇)中,将所得溶液喷涂到空白丸芯表面,直至达到目标增重,干燥、冷却,得到微丸A;b)微丸B(缓释层微丸)的制备:将不溶性薄膜材料、致孔剂以及可选的增塑剂溶解或混悬于适当的溶剂(比如:乙醇、二氯甲烷、丙酮)中,将所得溶液喷涂到主药层微丸(微丸A)表面,直至达到目标增重,干燥、冷却,得到微丸B;c)微丸C(速释药物层微丸)的制备:将琥珀酸美托洛尔和粘合剂溶解在适当的溶剂(比如:纯化水、丙酮、乙醇)中,将所得溶液喷涂到缓释微丸(微丸B)表面,直至达到目标增重,得到微丸C;微丸C可以与其他辅料一起压制成片剂或填入胶囊,也可进一步进行缓冲层的包衣,干燥、冷却,得到微丸D(缓冲层微丸);d)可选的微丸D(缓冲层微丸)的制备:将蜡质材料溶解在适当的溶剂(比如:纯化水和乙醇的混合溶剂、纯化水和丙酮的混合溶剂)中,将所得溶液喷涂到速释药物层微丸(微丸C)的表面,直至达到目标增重,干燥、冷却,得到微丸D;e)微丸C或微丸D均可以与其他辅料一起压制成片剂或填入胶囊。在上述制备方法中,可以用流化床包衣的方式喷涂各步骤中所得到的微丸。本发明与现有技术相比,具有如下有益效果:a)本技术采用蔗糖丸芯为起始丸芯,有多种可供选择的市售的商业化来源,且早已被美国药典收载,便于质量控制。b)本技术在包覆药物层之前无需再包覆一层额外的不溶性聚合物衣层。因为蔗糖丸芯不会溶胀,因此无需特殊包衣限制其溶胀。c)本技术在缓释层之外额外的包覆了一层速释药物层,使其即使在较高的载药量(>40%)时,在1h以内就能释放出不低于总剂量5%的药物,并且能达到美国药典的要求,可以迅速地发挥疗效,在前8h内呈现出直线式的释放曲线并维持20h以上。d)本技术制备的琥珀酸美托洛尔缓释微丸无需再与其他速释组分混合,不存在混合均匀性的问题,易于实现工业放大。附图说明图1说明了琥珀酸美托洛尔缓释微丸的结构示意图。图2说明了在实施例4中微丸B1和微丸C1,以及实施例7中片剂1按照美国药典方法测定的前8h的释放曲线。图3说明了在实施例5中微丸B2和微丸C2,以及实施例8中片剂2按照美国药典方法测定的前8h的释放曲线。图4说明了在实施例6中微丸B3和微丸C3,以及实施例9中片剂3按照美国药典方法测定的前8h的释放曲线。具体实施方式本领域技术人员应当理解下述实施例仅为说明的目的提供,不应以任何方式解释为对本公开的限制。实施例1:琥珀酸美托洛尔缓释微丸的制备处方:*工艺过程中除去制备工艺:1)将琥珀酸美托洛尔和羟丙甲纤维素溶解于纯化水中,将蔗糖丸芯置于流化床中,设置进风温度50~70℃,进风量70~120m3/h,雾化压力1.5~2.5bar,喷液速度6~20g/min。包衣液喷完之后,保持进风温度,干燥0.5~2h,即得微丸A1。2)将乙基纤维素和羟丙基纤维素溶解于乙醇中,将上一步制备得到的微丸A1置于流化床中,设置进风温度40~50℃,进风量70~120m3/h,雾化压力1.5~2.5bar,喷液速度3~16g/min。包衣液喷完之后,保持进风温度,干燥0.5~2h,即得微丸B1。3)将微丸B1投入流化床中进行速释药物层包衣,制备工艺同药物层包衣步骤1);即得微丸C1。4)将聚乙二醇溶解于乙醇和纯化水的混合溶剂中,将上一步制备得到的微丸C1置于流化床中,设置进风温度40~50℃,进风量70~120m3/h,雾化压力1.5~2.5bar,喷液速度5~20g/min。包衣液喷完之后,保持进风温度,干燥0.5~2h,即得微丸D1。所得微丸D1的载药量(活性药物在最终微丸中的重量比例)为59.6%。压片制备工艺:将微丸D1与微晶纤维素、交联羧甲基纤维素钠、二氧化硅和硬脂酸镁混合均匀后,压制成不同剂量规格的片剂1。实施例2:琥珀酸美托洛尔缓释微丸的制备处方:*工艺过程中除去制备工艺:1)将琥珀酸美托洛尔和聚乙烯吡咯烷酮溶解于丙酮中,将蔗糖丸芯置于流化床中,设置进风温度40~60℃,进风量70~120m3/h,雾化压力1.5~2.5bar,喷液速度6~20g/min。包衣液喷完之后,保持进风温度,干燥0.5~2h,即得微丸A2。2)将乙基纤维素和羟丙甲纤维素溶解于二氯甲烷中,将上一步制备得到的微丸A2置于流化床中,设置进风温度35~50℃,进风量70~120m3/h,雾化压力1.5~2.5bar,喷液速度3~16g/min。包衣液喷完之后,保持进风温度,干燥0.5~2h,即得微丸B2。3)将微丸B2投入流化床中进行速释药物层包衣,制备工艺同步骤1);即得微丸C2。所得微丸C2的载药量(活性药物在最终微丸中的重量比例)为61.5%。压片制备工艺:将微丸C2与微晶纤维素、交联羧甲基纤维素钠、二氧化硅和硬脂酸镁混合均匀后,压制成不同剂量规格的片剂2。实施例3琥珀酸美托洛尔缓释微丸的制备处方:*工艺过程中除去制备工艺:1)将琥珀酸美托洛尔和羟丙纤维素溶解于乙醇中,将蔗糖丸芯置于流化床中,设置进风温度40~60℃,进风量70~120m3/h,雾化压力1.5~2.5bar,喷液速度6~20g/min。包衣液喷完之后,保持进风温度,干燥0.5~2h,即得微丸A3。2)将乙基纤维素和聚乙二醇溶解于丙酮中,将上一步制备得到的微丸A3置于流化床中,设置进风温度35~50℃,进风量70~120m3/h,雾化压力1.5~2.5bar,喷液速度3~16g/min。包衣液喷完之后,保持进风温度,干燥0.5~2h,即得微丸B3。3)将微丸B3投入流化床中进行速释药物层包衣,制备工艺同步骤1);即得微丸C3。4)将聚乙二醇溶解于丙酮和纯化水的混合溶剂中,将上一步制备得到的微丸C3置于流化床中,设置进风温度40~50℃,进风量70~120m3/h,雾化压力1.5~2.5bar,喷液速度5~20g/min。包衣液喷完之后,保持进风温度,干燥0.5~2h,即得微丸D3。所得微丸D3的载药量(活性药物在最终微丸中的重量比例)为43.8%压片处方和工艺同实施例1,得到片剂3。实施例4:将实施例1制备的微丸B1和微丸C1按照美国药典方法,在500ml,pH6.8的磷酸缓冲液介质中用桨法50rpm测定其释放度,其结果如表1所示。在进行速释药物层包衣之后,1h的释放度从2%提高到了7%。从附图2所示的释放曲线中也可以看出,在缓释层外面再进行一层额外的速释层包衣,不仅可以使延迟释放的现象得到改善,在满足美国药典标准的同时,还能保持药物匀速释放,前8h的释放曲线接近于直线。表1实施例1中的微丸B1和微丸C1的按照美国药典方法测定的释放度实施例5:将实施例2制备的微丸B2和微丸C2按照美国药典方法,在500ml,pH6.8的磷酸缓冲液介质中用桨法50rpm测定其释放度,其结果如表2所示。在进行速释药物层包衣之后,1h的释放度从1%提高到了7%。从附图3所示的释放曲线中也可以看出,在缓释层外面再进行一层额外的速释层包衣,不仅可以使延迟释放的现象得到改善,在满足美国药典标准的同时,还能保持药物匀速释放,前8h的释放曲线接近于直线。表2实施例2中微丸B2和微丸C2按照美国药典方法测定的释放度实施例6:将实施例3制备的微丸B3和微丸C3按照美国药典方法,在500ml,pH6.8的磷酸缓冲液介质中用桨法50rpm测定其释放度,其结果如表3所示。在进行速释药物层包衣之后,1h的释放度从0%提高到了6%。从附图4所示的释放曲线中也可以看出,在缓释层外面再进行一层额外的速释层包衣,不仅可以使延迟释放的现象得到改善,在满足美国药典标准的同时,还能保持药物匀速释放,前8h的释放曲线接近于直线。表3实施例3中的微丸B3和微丸C3按照美国药典方法测定的释放度实施例7将实施例1制备的200mg规格的琥珀酸美托洛尔缓释片(片剂1)按照美国药典方法,在500ml,pH6.8的磷酸缓冲液介质中用桨法50rpm测定其释放度,其结果如表4所示。该片剂能在最初的1h里释放8%的琥珀酸美托洛尔,同时满足美国药典的要求。整个释放过程可以持续20h,如附图2所示。表4实施例1制备的片剂1(200mg)按照美国药典方法测定的释放度时间[h]美国药典接收范围释放度[%]1≤25%8420%-40%29840%-60%5820≥80%95实施例8:将实施例2制备的200mg规格的琥珀酸美托洛尔缓释片(片剂2)按照美国药典方法,在500ml,pH6.8的磷酸缓冲液介质中用桨法50rpm测定其释放度,其结果如表5所示。该片剂能在最初的1h里释放9%的琥珀酸美托洛尔,并同时满足美国药典的要求。整个释放过程可以持续20h,如附图3所示。表5实施例2制备的片剂2(200mg)按照美国药典方法测定的释放度时间[h]美国药典接收范围释放度[%]1≤25%9420%-40%29840%-60%5920≥80%91实施例9将实施例3制备的200mg规格的琥珀酸美托洛尔缓释片(片剂3)按照美国药典方法,在500ml,pH6.8的磷酸缓冲液介质中用桨法50rpm测定其释放度,其结果如表5所示。该片剂能在最初的1h里释放5%的琥珀酸美托洛尔,并同时满足美国药典的要求。整个释放过程可以持续20h,如附图4所示。表6实施例3制备的片剂3(200mg)按照美国药典方法测定的释放度时间[h]美国药典接收范围释放度[%]1≤25%5420%-40%24840%-60%5620≥80%90本领域技术人员应当理解,本发明技术方案中所涉及的数值或数值端点,其含义或意在保护的范围并不局限于该数字本身。本领域技术人员能够理解,它们包含了那些已被本领域广为接受的可允许的误差范围,例如:实验误差、测量误差、统计误差和随机误差等,而这些误差范围均包含在本发明的范围之内。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1